서 론
골격근 세포에서 Ca2+은 골격근 수축의 핵심 인자이며, 다양한 신호체계를 조절하는 핵심 인자 이다[1]. 골격근에서 세포내 Ca2+은 근형질세망(sarcoplasmic reticulum, SR), 미토콘드리아, 핵, 리보솜(lysosome) 등과 같은 세포내 소기관에서 유기적으로 방출되어, 일종의 Ca2+ 신호(oscillation)를 형성함으로써 Ca2+ 농도에 반응하는 다양한 단백질을 활성화시킨다[2,3]. SR과 미토콘드리아 사이를 SR (ER)-mitochondria junction이라고 특별하게 명명하고 있으며, 두개의 세포내 소기관이 주고 받는 Ca2+ 이동이 미토콘드리아 기능 및 항상성 유지에 중요함이 제시되고 있는데, 특히 에너지 대사 체계의 활성화 조절에 밀접한 관련이 있다고 하였다[4]. SR Ca2+은 ryanodine receptor (RYR), inositol-3-phosphate receptor (IP3R)와 같은 Ca2+ channel과 sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) 등에 의해 조절되며, SR Ca2+은 다양한 세포내 신호기전에 의해 조절될 수 있다[5,6]. 대표적으로 골격근 수축 초기 급격히 증가할 수 있는 해당과정(glycolysis)은 SR로의 Ca2+ 유입을 촉진할 수 있는 것으로 알려져 있는데, SR 표면에는 aldolase, GAPDH, phosphoglycerate kinase (PGK), phosphoglyceromutase, enolase, and pyruvate kinase (PK)와 같은 해당과정의 핵심적인 효소들이 존재하며, ATP 생성을 통해 SERCA을 활성화하여 SR로의 Ca2+ 유입을 증가시킨다[7]. 따라서 골격근 수축에 의해 나타나는 대사적 변화는 SR Ca2+을 조절하는 중요 조절자로 작용할 수 있으며, SR-미토콘드리아의 Ca2+ 상호작용이 미토콘드리아 생합성을 조절하는 중요한 요인으로 작용할 수 있을 것이다 그러나, 골격근 수축시 골격근에서 SR과 미토콘드리아 간의 Ca2+ 신호 상호 조절 연구에 관련된 연구는 미비한 실정이다. 반면, 미토콘드리아에는 Ca2+ 이온의 이동을 조절하는 두가지 중요한 이동통로(channel)가 존재하는데, 미토콘드리아 외막의 voltage dependent anion channel (VDAC)과 미토콘드리아 내막의 mitochondria calcium uniporter (MCU) 같은 Ca2+ channel이 발현되어 있으며, 미토콘드리아 내 Ca2+ 수준을 조절하는 기능한다[8]. VDAC 및 MCU는 미토콘드리아 Ca2+ 수준을 유기적으로 조절하여, 미토콘드리아 항상성을 유지시키며, α-ketoglutarate dehydrogenase와 같은 에너지 생성과 관련된 Ca2+ 의존성 에너지 대사 단백질 및 다양한 대사를 조절하는 중요 인자이다[9]. 미토콘드리아 Ca2+ channel을 조절하는 인자는 명확하게 밝혀져 있지 않는데, 최근 Ca2+/CaM activated protein kinase (CaMKII)가 미토콘드리아의 MCU를 활성화하는 것으로 보고 되고 있으며, 해당과정의 rate-limiting enzyme인 hexokinase 또한 VDAC과 결합하여, 기능하는 것으로 알려져 있으나 구체적인 기전은 밝혀져 있지 않다[10-12]. Hexokinase의 경우, 미토콘드리아 생합성 및 항상성 유지에 중요한 mitophagy 과정에서 관여되어 있을 뿐만 아니라[13], 미토콘드리아 Ca2+ 수준과 potential 유지에 중요한 역할을 하는 것으로 알려져 있다[14]. 따라서 근수축시 세포질에서 발생하는 에너지 대사과정은 미토콘드리아 Ca2+ 및 생합성 조절에 영향을 미칠 것으로 생각되며, 골격근에서 에너지대사의 변화는 세포질과 미토콘드리아 간의 에너지 대사 및 대사산물(단백질 및 기질-생성물 등)의 발현 및 변화에 따라 유기적으로 조절될 가능성이 있다. 따라서 근수축시 에너지 대사의 변화에 따른 세포내 SR과 다른 세포내 소기관인 미토콘드리아의 상호조절 기전에 대한 연구가 필요하다.
본 연구에서는 골격근에서 근세사(muscle fiber) 를 분리하여, 골격근 수축시 미토콘드리아 Ca2+ 수준 및 potential을 조절하는 세포내 대사과정에 대한 연구를 실시하였으며, 특히 근수축시 해당 과정의 억제가 SR의 Ca2+ 수준과 미토콘드리아의 Ca2+ 수준과 potential 변화에 어떠한 변화를 가져오는지를 규명하는 것을 연구의 목적으로 설정하였다. 이를 위하여, 2-deoxy-glucose와 3-bromo-pyruvate, 두가지 해당과정 억제제(inhibitor)를 사용하여 골격근 수축시 해당과정을 억제하였으며, 그에 따른 세포질과 미토콘드리아 Ca2+ 수준 변화와 SR Ca2+ 수준의 변화를 관찰한 결과, 근수축시 해당과정은 SR Ca2+ 수준과 미토콘드리아 Ca2+ 수준에 조절에 중요한 조절자임을 확인하였다.
연구 방법
1. 연구 대상
이 연구에 사용된 실험동물은 출생 시기(8주령)가 동일한 C57BL/6 mice를 이용하였으며, mice는 J실험동물에서 구입하였다. 실험동물의 생육환경은 Specific-pathogen-free (SPF) system에서 12시간 주기의 명암과, 20℃ 그리고 습도는 40% 수준을 유지하였다. 모든 실험동물의 사육 및 실험은 Guide line for care and use of laboratory animal의 국제표준을 준수하였다. 동물실험과 관련된 연구윤리심의는 C 대학의 동물실험윤리위원회의 심사를 시행하였다(CBU 2016-79).
2. 연구절차
1) Skeletal muscle single fiber 분리
골격근에서 Single fiber를 분리하기 위해 각 실험동물은 경추 탈골을 실시하여, 비복근(gastrocnemius)을 적출하였다. 분리된 비복근 중 Red gastrocnemius portion을 이용하였으며, 이 후 37℃로 미리 준비된 0.2% type I collagenase (CLS-1, Washington, USA)가 들어가 있는 dulbecco’s Modified Eagle’s medium (DMEM) buffer (LM 001-05, Wellgene, Korea)에 적출된 골격근을 넣고 37℃ shaking water bath에서 2시간 동안 80-90 RPM으로 incubation 하였다. 효소를 통한 분리가 끝나고 single fiber가 분리되면 single fiber가 포함된 DMEM buffer를 10% horse serum (26050-088, Thermo Fisher, USA)와 10% FBS buffer (s101-01, Welgene, Korea)이 포함된 DMEM로 교환하여, Petri dish에 옮기고 37℃ CO2 incubator에서 overnight incubation을 실시하였다. 2일차에 Pasture pipette을 이용하여 single fiber를 분리하고 다시 37℃ CO2 incubator에서 overnight incubation을 실시하였다. Collagenase로 분리된 세포는 1-2일차의 경우, 세포내 Ca2+ 신호로 인한 변화를 관찰하기 어렵다는 Backer et al. [15]의 연구를 참조하여, 분리된 세포는 3일차에 실험에 이용되었으며, 실험 전까지 37℃ CO2 incubator에 보관하였다.
2) 골격근 primary cell의 전기자극(골격근 수축 simulation)
전기자극과 관련된 실험 프로토콜은 Liu et al. [16]이 이용한 방법을 사용하였다. Mouse에서 분리되어 DMEM medium buffer에 보관되어 있던 single fiber는 krebs ringer buffer (115 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 1.2 mM NaH2PO4, 1.2 mM Na2SO4, 2.5 mM CaCl2, 25 mM NaHCO3, 10 mM glucose, pH 7.4)로 옮겨지게 되고 1.5 mL 튜브 한 개당 muscle fiber를 100-120개씩 분주하였다. 37℃ 수조에서 5분 동안 안정을 취한 다음, 상온에서 carbon 소재의 전극을 사용하여, 전기자극기(DS2A mk2, Digitimer, England)로 자극을 실시하였다. 자극에 필요한 저주파 주파수는 10 Hz로 하고, 자극이 주어지는 시간은 1초, 자극 간 간격은 1초로 실시하였다. Ca2+ 측정 및 단백질 인산화 수준 변화 측정을 위한 전기자극의 지속시간은 300초를 기준으로 실시하였다.
3) Confocal micro scope를 이용한 골격근 세포에서 Ca2+ 변화 측정
전기자극에 의한 single fiber 수축시 세포내 및 각 소기관의 Ca2+ 변화를 측정하기 위해 각기 다른 Ca2+ dye를 이용하였다. Flou-4 AM (F14201, Thermo Fisher, USA)을 이용하여, 세포내 Ca2+을 측정하였으며, SR Ca2+은 flou-5N (F14204, Thermo Fisher, USA) 그리고 미토콘드리아 Ca2+은 Rhod-2 AM (R1245MP, Thermo Fisher, USA) 시약을 이용하여 측정하였다. CO2 incubator에 보관되어 있던 single fiber는 30-40개씩 confocal dish로 분주되며, 50 nM의 Ca2+ dye가 포함된 DMEM buffer로 교체된 후 약 30분 동안 incubation되었다. 이후 새로운 DMEM buffer를 이용하여, 세포 안에 흡수되지 않은 Ca2+ dye 시약이 제거되며, 이후 컨포컬 현미경(Confocal microscope, Nikon C1, Nikon, Japan)에서 전기자극을 실시하면서 fluorescence 값을 측정하였다. Tsien et al. [17]에 의한 세포내 Ca2+ 산정 공식 [Ca2+]i=Kd(F-Fmin)/(Fmax-F)에 의거하여 세포내 Ca2+ 수준을 판정하였으며, flou-4에 대한 Kd 값은 335 nm, F 값은 측정되는 flourescense 값을 이용하였다. 측정이 끝난 후, Fmax는 8 μM의 ionomycin을 처치하여 측정하였으며, Fmin은 50 mM의 EGTA를 처치하여 값을 측정하였다. SR 및 미토콘드리아 Ca2+은 상대적인 fluorescence 값을 비교하여 측정하였다. 측정에 이용되는 excitation/emission 값은 480/520 및 495/515 nm를 사용하였다.
4) Confocal micro scope를 이용한 골격근 세포에서 mitochondria membrane potential 변화 측정
Glycolysis inhibitor에 의한 mitochondria membrane potential 변화를 측정하기 위해 TMRM을 이용하였다. CO2 incubator에 보관되어 있던 single fiber는 30-40개씩 confocal dish로 분주되며, TMRM (T668, Thermo Fisher, USA) 50 nM이 함유된 DMEM buffer에 약 30분 동안 incubation되었다. 이후 컨포컬 현미경(Confocal microscope, Nikon C1, Nikon, Japan)에서 100 μM carbonyl cyanide m-chlorophenylhydrazone (CCCP)를 처치하면서 fluorescence 값을 측정하였다. CCCP를 처리한 후 최고점(high)의 fluorescence 값에서 최저점(low) 간의 차이를 membrane potential로 간주하여 측정하였으며, 최고점-최저점을 값이 높은 시료가 상대적으로 membrane potential이 높음을 나타내었다. 측정에 이용되는 excitation/emission 값은 548/574 및 495/515 nm를 사용하였다.
5) Single fiber에 사용된 Ca2+ channel 억제제 및 glycolysis 억제제 용량 및 용법
Single fiber에 사용된 Ca2+ channel 억제제 및 glycolysis 억제제 용량 및 용법은 다음과 같다. 2-deoxy glucose (2-DG, 5 mM Sigma Aldrich, USA), 3-bromo pyruvate (3BP, 100 μM, Sigma Aldrich, USA). Thapsigargin (Thap, 100 μM, Sigma Aldrich, USA). 일반적으로 처치 시간은 Ca2+ 측정 전 골격근 세포에 10분 정도 처치한 후 측정을 실시하였다.
연구 결과
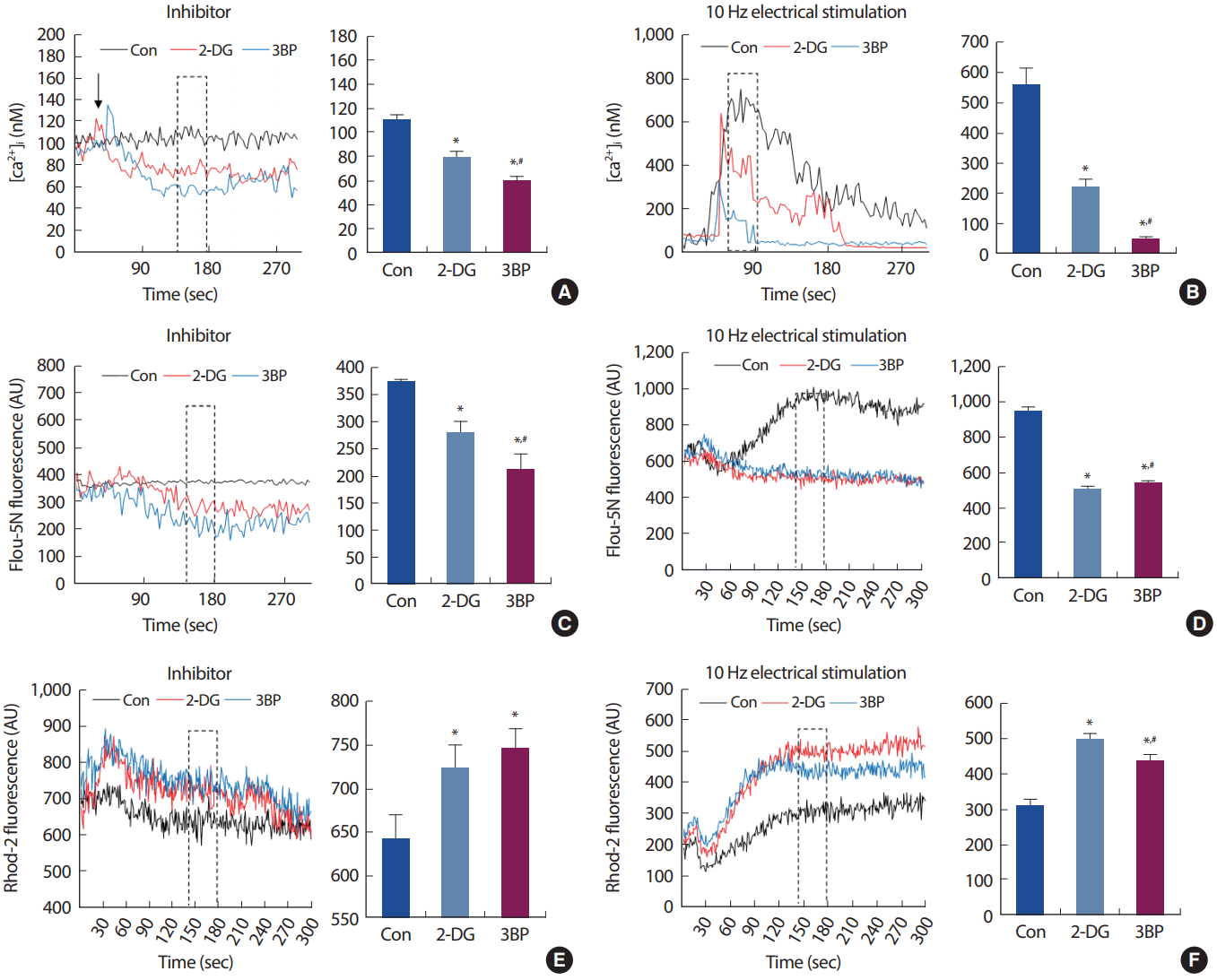
1. 해당과정 억제제(glycolysis inhibitor) 처치에 따른 골격근 single fiber 에서 근수축시 세포내 Ca2+ 수준의 변화
근수축시 glycolysis inhibitor가 세포내 Ca2+에 어떠한 영향을 미치는지를 알아보기 위해 근세사에 해당과정 억제제를 사전 처치한 후 세포내 Ca2+의 변화를 관찰하였다. 골격근 세포내(cytosolic) Ca2+은 각 억제제 처치시 대조군(Con)에 비해 유의하게 감소하는 것으로 나타났으며, 억제제 사전처치 후 근수축에 의한 세포내 Ca2+ 증가도 대조군에 비해 유의하게 감소하는 것으로 나타났다(Fig. 1A, B). Glycolysis inhibitor 처치가 SR Ca2+ 수준에 미치는 영향을 측정하기 위하여, SR Ca2+ fluorescence dye인 Flou-5N을 이용하여 SR Ca2+ 변화를 측정하였다. Glycolysis inhibitor 처치는 골격근 세포의 SR Ca2+ 수준을 대조군보다 유의하게 감소시켰으며, 억제제 사전 처치 후 골격근 수축시 증가하는 SR Ca2+ 수준의 변화도 대조군보다 유의하게 감소하는 것으로 나타났다(Fig. 1C, D). 미토콘드리아 Ca2+ 수준은 Rhod-2 AM을 이용하여 측정하였으며, glycolysis 억제제 처치 결과, glycolysis inhibitor는 미토콘드리아 Ca2+을 대조군보다 증가시키는 것으로 나타났으며, 억제제 사전 처치 후 골격근 수축시에도 대조군보다 유의한 증가를 나타내었다. 이러한 결과는 glycolysis inhibitor가 안정시 세포내 Ca2+수준을 감소시킬 수 있음을 제시하고 있다. 반면 미토콘드리아 Ca2+ 수준은 증가하였음으로 glycolysis inhibitor는 미토콘드리아에 Ca2+을 과부화시키는 효과를 가져올 수 있음을 제시하고 있으며, 전체적인 세포내 Ca2+ 수준의 변화는 주로 SR의 Ca2+ 수준의 감소로 인한 것임을 알 수 있다.
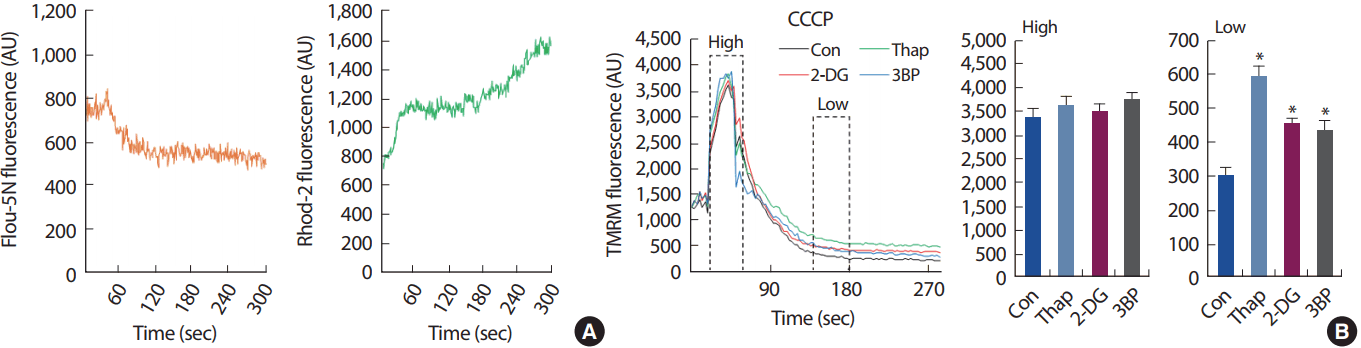
2. 해당과정 억제제(glycolysis inhibitor) 처치에 따른 골격근 single fiber 에서 미토콘드리아 membrane potential 수준의 변화
안정시 및 근수축시 glycolysis inhibitor가 SR Ca2+ 수준을 감소시키는데 영향을 미치고 있음을 확인하였음으로, SR Ca2+ 수준을 인위적으로 감소시킬 경우, mitochondria Ca2+ 수준이 변화하는지를 실험하였다. Sarcoplasmic/Endoplasmic reticulum ATPase (SERCA) inhibitor인 Thapsigargin은 SR Ca2+ pump인 SERCA를 억제함으로써 SR의 Ca2+을 고갈(depletion) 시키는 것으로 알려져 있다[18]. 골격근 세포에 thapsigargin (Thap)을 처치한 결과, SR Ca2+이 고갈되고 있음을 확인하였으며, 이와 함께 mitochondria Ca2+이 증가되는 것을 확인하였다(Fig. 2B). 미토콘드리아의 Ca2+ 과부하는 mitochondria membrane potential (MMP)을 억제할 수 있다고 알려져 있으며 이는 미토콘드리아 에너지 생성을 저해하는 요인으로 작용할 수 있기 때문에 골격근에 thapsigargin, glycolysis inhibitor를 처리하고 MMP를 측정하였다[19]. MMP는 TMRM을 이용하여 측정하였으며, 100 μM의 CCCP 처치에 의한 변화를 관찰하였다. Glycolysis inhibitor와 thapsigargin 처치는 골격근 세포의 MMP를 대조군에 비해 유의하게 감소시키는 경향을 보였다(Fig. 2B). 이러한 결과는 glycolysis inhibitor이나 thapsigargin 처리에 의한 미토콘드리아 Ca2+ 과부하가 MMP를 낮추는 주요원인임을 제시하고 있다.
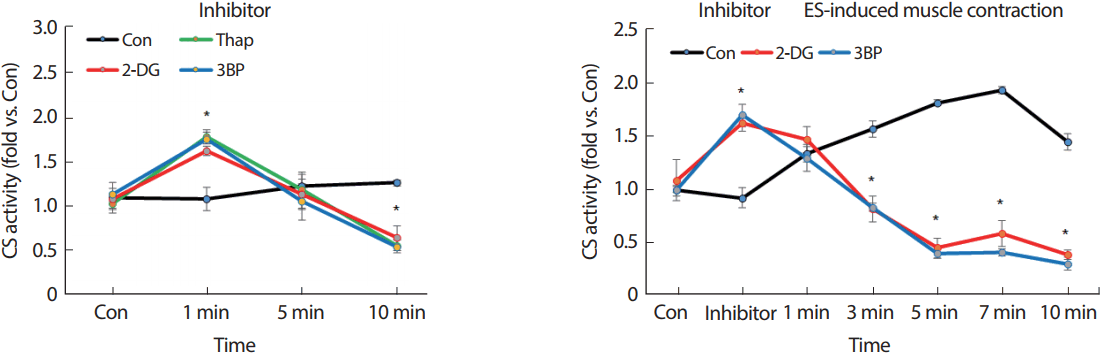
3. 해당과정 억제제(glycolysis inhibitor) 처치에 따른 Citric acid synthase (CS) activity의 변화
Glycolysis inhibitor 및 thapsigargin 처치에 의한 미토콘드리아 Ca2+ 과부하가 MMP 감소의 원인이 됨을 확인하였으므로, 억제제 사용에 따른 안정시 및 근수축시 미토콘드리아 에너지 생성 기전에 중요한 citric acid cycle의 조절 단백질인 CS의 활성을 관찰하였다. Glycolytic inhibitor와 thapsigargin을 골격근 세포에 처리한 결과, 처리 후 1분에 대조군 보다 유의한 증가를 관찰하였으며 처리 후 10분에는 대조군 보다 유의한 감소를 나타내었다. 이러한 결과는 glycolysis inhibitor에 의해 미토콘드리아 Ca2+ 과부하가 빠르게 증가하며(Fig. 2B), 이로 인한 미토콘드리아 내 증가한 Ca2+ 수준이 에너지 생성 체계에 영향을 미치고 있음을 제시하고 있다(Fig. 3A). Glycolysis inhibitor를 사전 처치 후 골격근 수축을 실시한 결과에서도 CS activity가 유의하게 감소하는 경향을 나타내었는데, 이러한 결과는 골격근 수축시 glycolysis에 의한 미토콘드리아 Ca2+ 수준 조절이 미토콘드리아 에너지 생성에 중요한 요인임을 제시하고 있다(Fig. 3B).
논 의
골격근에서 미토콘드리아는 근수축을 위한 에너지 생성에 중요한 역할을 할 뿐만 아니라, autophagy, inflammation, muscle type turn over와 같은 다양한 세포내 대사 과정과 연관되어 있다[20]. 특히 미토콘드리아 Ca2+은 미토콘드리아 metrix의 calcium dependent protein, α-ketoglutarate dehydrogenase 등과 같은 에너지 대사 관련 단백질의 활성을 조절하는 중요 인자이며, 이를 통해 근수축에 필요한 에너지를 조절하는 중요한 기능을 하고 있다[9]. 그러나 미토콘드리아의 Ca2+이 과부화 되면 세포 항상성(homeostasis)가 저해되며, 근위축(myophthy), 근손상, 당뇨병 등의 다양한 질병의 원인이 나타나기도 한다[21]. 본 연구에서는 SR의 Ca2+ 수준이 미토콘드리아 Ca2+ 수준을 조절하는 중요 기전이라는 사실을 바탕으로 골격근에서 근수축시 해당과정의 억제가 미토콘드리아 Ca2+ 수준에 어떠한 영향을 미치는지를 규명하는 것이 목적이었다[22]. 그 결과 해당과정은 근수축시 SR Ca2+ 증가 및 유지하는데 관여되어 있음을 확인하였으며(Fig. 1C, D), 이는 미토콘드리아 Ca2+ 조절에도 관여되어 있을 가능성을 제시하고 있다. 해당 과정과 관련된 Ca2+ 신호 기전에 의해 미토콘드리아 Ca2+ 신호가 조절될 수 있으며, 이는 SERCA의 활성 조절에 의한 세포질의 Ca2+ 수준이 중요한 조절 기전 임을 밝히고 있는 것이다.
모든 세포에서 Ca2+ 신호 기전은 필수적인 요소로써 다양한 세포내 대사를 조절하는 것으로 알려져 있다[23]. 특히 SR과 근접한 mitochondria 간의 연접부를 SR-mitochondria junction이라고 명명하며, 미토콘드리아 Ca2+ 수준을 조절하는 중요 기제로 보고하고 있는데, 당뇨병, 근손상, 근섬유 전환 등의 다양한 대사 조절의 핵심요소로 제시되고 있다[24,25]. 이러한 연접부를 구성하고 조절하는 tethering protein으로써는 GRP-75 등이 제시되고 있으며, VDAC, IP3 receptor와 같은 Ca2+ channel 또한 인접해 있는 것으로 보고 되고 있다[26]. 그러나 이들 두 기관과의 Ca2+ 수준 상호 조절 기전에 대해서는 명확히 밝혀지지 않았으며, 현재 많은 연구가 진행 중에 있다.
본 연구에서는 glycolysis inhibitor를 이용하여, 근수축시 발생하는 미토콘드리아 Ca2+ 수준의 변화를 관찰하고자 하였다. 이와 관련하여 Panfili & Sandri [27]의 연구에서는 해당과정의 중요 rate-limiting kinase인 hexokinase가 세포내 미토콘드리아의 Ca2+ 수준을 조절할 수 있는 중요 조절 장치임을 제시한 바 있다. 최근의 연구에서는 hexokinase가 미토콘드리아 외막의 Ca2+ channel인 VDAC의 활성을 조절함으로써 미토콘드리아 Ca2+ 수준을 조절할 수 있다고 제시되고 있으며, 이를 통해 세포질에서 나타나는 에너지 대사 체계의 변화와 그에 의한 다양한 단백질의 변화가 미토콘드리아 Ca2+ 조절에 중요한 역할을 할 수 있다고 하였다[14]. 본 연구에서는 glycolysis inhibitor 처치시 SR Ca2+ 수준의 감소와 함께, 미토콘드리아 Ca2+ 수준이 증가하는 것을 확인하였다(Fig. 1C, E). 이러한 결과는 glycolysis inhibitor 처치시 SERCA의 활성 저하로 인하여, SR Ca2+이 점차 고갈되고 이로 인하여 세포질의 Ca2+ 증가와 미토콘드리아 Ca2+ 증가가 나타난 결과로 생각된다. Glycolysis는 SERCA 활성을 조절하는 중요 인자로 보고되고 있으며, 이는 근수축을 지속하기 위한 기제로써 작용한다[28]. SR 표면에는 glycogenolytic/glycolytic enzyme이 존재할 뿐만 아니라, creatine kinase도 발현되어 있으며, pyruvate kinase, aldolase 그리고 glyceraldhyde-phosphate dehydrogenase와 같은 에너지 생성 관련 단백질이 SERCA 주변에 존재하여 ATP 생성을 통한 SERCA 활성 조절 역할을 한다[29,30]. 따라서 근수축 동안의 해당과정의 활성화는 SERCA 활성 조절을 위한 ATP 생성의 기제로써 작용할 수 있으며, 이는 근세포의 Ca2+ 항상성 유지와 미토콘드리아 Ca2+ 수준의 유지에 중요한 역할을 할 수 있다. 반면 본 연구에서와 같이 glycolysis inhibitor를 사용한 경우, 해당과정에 의한 ATP 생성이 제한되어 SERCA 활성을 저해할 수 있으며, 이는 본 연구의 Fig. 2A와 같이 SERCA inhibitor인 thapsigargin을 사용한 결과와 같이 SR Ca2+ 수준을 저해할 가능성이 있을 것으로 생각된다.
SERCA 억제에 의한 세포질 내의 Ca2+ 수준 변화는 미토콘드리아 Ca2+을 증가시키는 원인이 된다[31]. SERCA의 억제는 ryanodine receptor (RYR)로부터의 Ca2+ leak을 증가시키며, L-type Ca2+ channel의 활성화를 통해 세포질의 Ca2+ 수준을 증가시키는 요인이 된다[32]. 증가된 세포질 내 Ca2+은 미토콘드리아의 활성을 증가시키기도 하지만 세포의 에너지 생성을 저해하기도 하는데[33], 이는 미토콘드리아 Ca2+이 과부하되어 나타나는 현상이다[34,35]. 본 연구에서도 glycolysis inhibitor는 미토콘드리아 Ca2+ 수준을 증가시켰을 뿐만 아니라, 근수축시에는 대조군보다 더 높은 수준의 미토콘드리아 Ca2+을 나타내었는데, 이는 미토콘드리아 Ca2+ 과부하에 의한 MMP의 감소의 원인이 되었을 가능성이 있다. 따라서 본 연구의 Fig. 2와 3에서 나타난 glycolysis inhibitor에 의한 미토콘드리아 에너지 생성 능력의 기능 저하는 골격근에서의 과도한 Ca2+이 미토콘드리아로 이동하여 발생하였을 것으로 생각된다. Madreiter-Sokolowski et al. [36]에 의하면 미토콘드리아 Ca2+은 SERCA inhibition에 의한 세포질 Ca2+ 상승의 영향을 받으며, 미토콘드리아 ATP 생성의 영향을 받아 조절된다는 연구결과도 있다[37,38]. 이러한 결과는 미토콘드리아의 Ca2+ 수준이 SR Ca2+의 직접적인 조절을 받을 수도 있는 반면, SR Ca2+을 조절할 수 있는 가능성도 제공하고 있다. 그러나 glycolysis inhibitor 처치는 mitochondria Ca2+ level에 영향을 줄 수 있는 ROS, pH 증가의 원인이 될 수 있으며[39,40], glycolysis의 중간 매개물인 glucose-6-phosphate와 같은 경우, 증감에 따라 미토콘드리아 에너지 생성체계를 조절할 수 있는 가능성이 있다[41]. 따라서 미토콘드리아의 Ca2+ 수준을 조절하는 다양한 세포내 대사와 연관하여 고찰할 필요가 있다.
Glycolysis는 인체의 다양한 질병과 연관되어 있다. 암세포(cancer cell)의 경우, 일반적인 세포에 비해 해당과정에 의한 에너지 생성률이 10배 정도 증가되어 있으며, 허혈(hypoxia)와 같은 현상에도 증식을 위해 활발한 대사활동을 지속한다[42]. Glycolytic inhibitor는 암세포의 미토콘드리아 항상성에 영향을 미칠 뿐만 아니라, TCA cycle과 관련된 여러 효소의 활성을 저해함으로써 항암효과를 나타낼 수 있다[43,44]. 이러한 효과는 세포의 세포질에서의 해당과정과 미토콘드리아 대사가 서로 밀접한 관련이 있음을 보여주는 결과로써 임상적으로 해당작용이 미토콘드리아를 통한 세포 항상성 유지에 매우 중요한 변인 임을 제시하고 있다. 또한 pyruvate kinase deficiency와 같이 유전적으로 해당과정에 문제가 있는 경우 용혈성 빈혈(hemolytic anemia)와 같은 증상을 가져오기도 하는데, 적혈구(red blood cell)의 생성과정과 생성된 세포의 해당작용이 억제되면서 미토콘드리아 대사의 불균형을 통해 세포 항상성이 저해되는 결과를 나타낸다[45]. 운동 중 영양적 불균형은 운동선수의 용혈성 빈혈을 초래한다고 알려져 있는데[46], 철분 뿐만 아니라 해당과정 관련 에너지 체계의 변화와 그로 인한 미토콘드리아 항상성과의 관계에 영향을 미치는 다양한 변인에 대한 연구가 필요하다.
골격근의 경우, 근수축을 하는 동안 에너지 연속체의 개념으로 glycolysis에서 oxidative phosphorylation으로 전환되며, 세포질에서 미토콘드리아로의 에너지 생성이 전환되는 특이적 특성을 가지고 있다[47]. 따라서 근수축 동안 발생하는 에너지 대사의 생성물 및 활성화되는 protein kinase의 변화에 따른 SR-mitochondria Ca2+ 수준 조절 기전에 대한 추가적인 연구가 필요할 것으로 생각된다. 실제로 골격근 수축으로 인하여 활성이 조절되는 Ca2+/CaM activated protein kinase (CaMKII)의 경우, 미토콘드리아의 MCU이나 SR Ca2+ channel인 RYR1의 조절하는 중요 조절자로 알려져 있으며[10,48], LETM1과 같은 calcium uniporter 등도 SR Ca2+과 관여되어 있으나[49], 골격근에서의 연구가 부족한 실정이다. 차후 추가적인 연구를 통해 근수축시 에너지 대사에 의한 mitochondria Ca2+ 수준을 조절하는 단백질 및 channel에 대한 연구가 in vivo 및 in vitro 실험을 통해 밝혀져야 할 것이다.
결 론
미토콘드리아는 세포내 ATP 생성을 조절하는 중요 조절기관으로 근수축시 다양한 대사과정에 중요한 조절기제로써 많은 연구들이 진행 중이다. 미토콘드리아 Ca2+은 에너지 대사과정을 조절하는 핵심인자로써 에너지 대사 과정의 활성화/비활성화의 원인이 된다. 본 연구는 근수축시 glycolysis 과정의 억제가 미토콘드리아 Ca2+ 수준에 어떠한 영향을 미치는지를 규명하기 위하여, glycolysis inhibitor을 사용한 실험을 진행하였으며, SERCA에 의한 SR의 Ca2+ 수준이 미토콘드리아의 활성화 조절의 기전으로써 작용할 수 있음을 제시하고 있다. 이러한 결과는 운동을 지속하기 위한 에너지 생성과정에서 Ca2+ 신호 기전이 어떠한 역할을 하고 있는지를 제시하고 있는 것과 동시에 SR-mitochondria junction의 Ca2+ 수준 변화의 중요함도 알려주고 있다.
Glycolysis inhibitor는 골격근 수축시 SR Ca2+ 수준은 낮추는 반면, 미토콘드리아의 Ca2+ 수준은 대조군에 비해 증가하는 과부하 현상이 나타남을 보여주었다. 미토콘드리아 Ca2+ 과부하는 오히려 미토콘드리아 내부의 에너지 생성 관련 효소의 활성을 낮추는 결과를 보였으며, 이는 골격근 수축시 과도한 운동, 지속된 운동이나 질병이 있을 경우 등에서 미토콘드리아 Ca2+이 과부하되어 나타날 수 있는 현상의 기제로써 설명될 수 있으며, 근수축시 예상할 수 있는 에너지 연속체의 개념에서 세포질과 미토콘드리아가 에너지 생성 기전을 상호 조절하는 기제로써 예상해 볼 수 있을 것이다.
그러나 본 연구에서는 in vitro 실험을 통한 간접적인 연구결과만을 도출하였을 뿐만 아니라, SR-mitochondria junction의 조절단백질과 Ca2+ channel에 대한 연구는 실시하지 못하였음으로 추가적인 연구가 필요할 것이다.