INTRODUCTION
The largest group of transmembrane receptors is the superfamily of G protein-coupled receptors (GPCRs) [1]. These receptors are involved in numerous physiological events and represent the class of proteins most often targeted for pharmaceutical interventions [2,3]. It has been demonstrated that GPCRs play a crucial role in cardiovascular homeostasis under physiological and pathological circumstances [4,5]. Exaggerated activation of GPCRs including adrenergic receptors, angiotensin II receptors, or endothelin receptors ubiquitously expressed in vascular smooth muscle cells (VSMCs) or cardiac muscle results in hypertension or heart failure [6]. Hyper-contractility of VSMCs or cardiomyocytes by GPCR ligands (e.g. norepinephrine, angiotensin II), a major hallmark of cardiovascular disorder, causes blood pressure elevation and ventricular hypertrophy [7]. Given their importance in cell signaling, the activation of GPCR and subsequent intracellular signaling events are likely to be tightly regulated to ensure appropriate cellular function. Considerable progress in the understanding of regulators of G protein signaling (RGS) proteins has occurred during the last decade and the proteins have been reported to modulate GPCR-mediated signaling. Impaired regulation of GPCR signaling cascades is likely coupled to many cardiovascular disorders. It has been demonstrated that altered expression or dysfunction of RGS proteins results in hypertension and cardiac hypertrophy [8]. However, potential role of exercise intervention on RGS protein-mediated regulation of GPCR signaling has not been investigated. In this review, we will describe 1) characteristics of RGS protein, discuss 2) the role of RGS proteins in the cardiovascular system, and propose 3) potential impact of exercise intervention on RGS protein regulation.
CHARACTERISTICS OF RGS PROTEINS
RGS proteins as GTPase-activating proteins
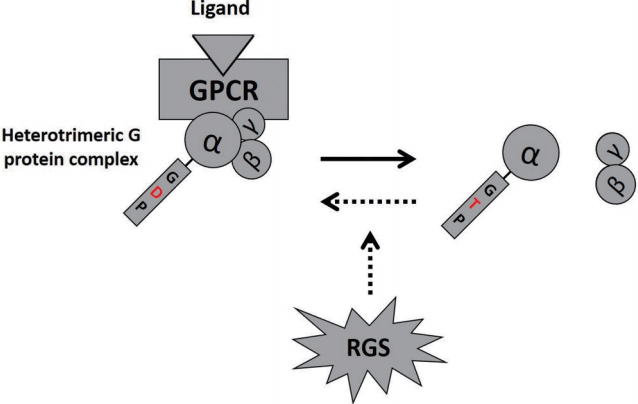
GPCR coupled heterotrimeric G proteins are comprised of α, β, and γ subunits and function as signal transducers. The trimeric G protein family is categorized into four classes based on the structure and function of the α subunit: 1) Gαs or 2) Gαi/o protein activate or inhibit adenylyl cyclase respectively, 3) Gαq protein stimulates various isoforms of phospholipase C (PLC) and in turn generates diacylglycerol (DAG) and inositol trisphosphate (IP3) by cleaving phosphatidylinositol bisphosphate (PIP2), and 4) Gα12/13 protein activates Rho GTPase. Ligand binding to an appropriate GPCR promotes exchange of GDP to GTP on the α subunit. The GTP-bound α subunit dissociates from β and γ subunit complexes and participates in downstream signaling cascades [9]. The magnitude and/or duration of the signaling is determined by how long the Gα subunit is activated, which is determined by the intrinsic GTPase activity of the α subunit. GTP hydrolysis by the intrinsic GTPase results in a conformational change of the α subunit, allowing re-association of GDPbound inactive α subunit with the β and γ subunit complexes. Thus the intracellular signaling response is terminated (Fig. 1). However, as the rate of intrinsic GTPase is relatively slow, a GTPase activating protein (GAP) is necessary for effective termination of G protein-mediated signaling. It is well accepted that RGS proteins serve as GAPs and fine-tune the GPCR activities by accelerating the GPTase activity up to 2,000 times [10]. Thus, GPCR-mediated signaling is controlled by regulating the rapid ‘on’-‘off’ kinetics of downstream effectors and diminishing sensitivity of GPCR [11].
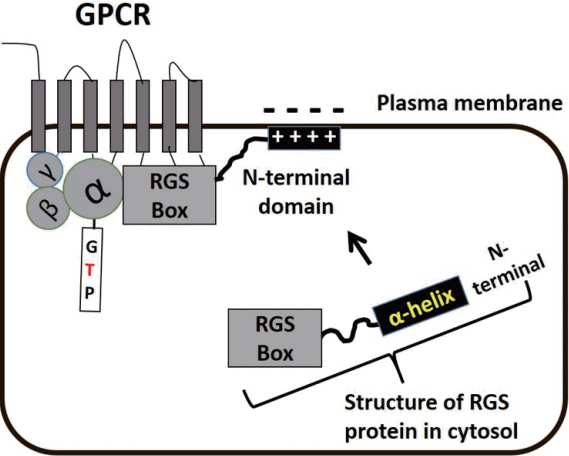
To date, more than 30 RGS proteins have been identified and classified based on their sequence homology and additional domains [12]. Of particular relevance, the RGS protein subfamily, B/R4, consisting of RGS1, 2, 3, 4, 5, 8, 13, 16, 18, and 21 proteins [13] is highly expressed in cardiovascular tissues [14]. Notably, RGS2, 4, and 5 proteins are widely distributed in heart and blood vessels and genes for the three proteins are located on the same chromosome and associated with blood pressure homeostasis [11]. The structure of B/R4 RGS proteins are commonly characterized by a core RGS carboxyl-terminal domain referred to as the ‘RGS box’ (Fig. 2). This region of the protein is composed of approximately 120 amino acids conferring the catalytic function of the RGS protein. Once GPCRs are activated, RGS proteins distributed in cytosol are translocated towards to GPCRs on the plasma membrane. The RGS box directly interacts with Gα protein of the GPCR, regulating GTPase activity and modulating G protein-dependent intracellular signaling (Fig. 2). While the N-terminal domain (33 amino acids) of RGS proteins does not take part in the acceleration of GTPase, the amphipathic α helical structures of this domain provide sites for interaction with the plasma membrane [15,16] (Fig. 2). However, only RGS3 protein interestingly has PDZ (PSD95/Dlg/Z0-1/2)-PEST (proline, glutamin, serine, threoninerich)-acidic N-terminal domain, instead of the amphipathic α helical structure shown in other B/R4 RGS proteins [13].
Selective regulation of RGS proteins
RGS proteins appear to have functional similarity for the modulation of GPCR activity in the cardiovascular system. It has been suggested that individual RGS proteins of the B/R4 subfamily indiscriminately interacts with Gαi or Gαq/11 protein [9]. However, since a variety of GPCRs and RGS proteins are not evenly expressed in all cells or tissues, it has been recently considered that RGS proteins may differentially interact with specific GPCRs. Therefore, numerous investigations have tested the hypothesis that RGS proteins preferentially and precisely discriminate specific GPCR bound to the same Gα protein [17].
Studies have demonstrated that RGS3 protein selectively inhibits endothelin-1 receptor-mediated signaling while RGS1 and 2 proteins show negligible inhibitory effects on the endothelin-1 receptor signaling pathways [18]. In that study, substantial attenuation of angiotensin II type 1 receptor (AT1R)-dependent signaling is observed in a manner dependent on RGS2 and 3 proteins, but not RGS4 protein. Additionally, RGS4 protein efficiently attenuates Ca2+ signaling in response to activation of cholinergic receptors in pancreatic acinar cells [19]. In particular, cholinergic receptors are preferentially regulated by RGS4 protein. In glomerular mesangial cells, urotensin II-mediated elevation in intracellular global Ca2+ levels is specifically modulated by RGS2 protein [20]. In vascular smooth muscle cells (VSMCs), RGS3 or 5 protein is involved in selectively control of signaling of the acetylcholine receptor or AT1R, respectively [21]. With respect to B/R4 RGS proteins outside of the cardiovascular system, RGS8 and 16 proteins have been reported to selectively interact with M1 muscarinic receptor expressed in xenopus oocytes or alpha2-adrenergic receptor in COS-7 fibroblast-like cell line, respectively [22,23]. CXCL12 (chemokine protein) and adenosine-activated GPCR signaling is regulated by RGS13 in human mast cells [24]. While RGS18 and 21 proteins predominantly expressed in osteoclasts [25] and taste bud cells [26], respectively, regulate GαqPCR, their specific selectivity has not been fully delineated. General characteristics of B/R4 RGS proteins are summarized in Table 1 [13,17].
The N-terminal domain of RGS proteins provides specificity of the RGS protein. The ability of RGS4 protein, deleted of its N-terminal domain, to inhibit Gαq protein-induced Ca2+ signaling is approximately 10,000 times less than the full-length RGS4 protein [27]. Similarly, N-terminal mutation decreases the inhibitory effects of RGS5 protein on Ca2+ signaling in response to application of Ang II and endothelin-1. Further, the N-terminal-deleted RGS5 protein is largely confined to the cytosol despite agonist stimulation [28]. Moreover, the same research group has compared abilities of RGS2, 4, and 5 proteins to regulate AT1R-mediated Ca2+ signaling [29]. As a consequence, it has been found that inhibition of Ca2+ signaling is dependent on the total volume of RGS proteins transfected to HEK-293 cells. Among three RGS proteins, RGS2 protein is a potent modulator for AT1R signaling. Notably, switching of the N-terminal domain between RGS2 and 5 proteins dramatically alters their inhibitory effects on downstream pathways following AT1R activation [29], further confirming that the selectivity of RGS proteins to specific GPCR may be determined by the properties of their N-terminals.
Scaffolding proteins for regulation of RGS proteins
The conserved RGS domain (so-called ‘RGS box’) is an important region where Gα subunit is bound and GTPase activity on the subunit takes place [30]. However, the exact mechanisms underlying recognition of RGS proteins by GPCR remains uncertain. In regard to the fundamental question of how RGS proteins identify GPCR, various scaffolding proteins including homer-2, neurabin, 14-3-3, or spinophilin have been investigated. In particular, spinophilin is highly expressed in neuronal dendritic spines and brain tissues (brainstem, hypothalamus, and cerebellum) [31,32]. A fascinating hypothesis has been proposed that spinophilin acts as a linker protein connecting between RGS proteins and GPCR [33]. These investigators have examined Ca2+ signaling responses to epinephrine (10 & 100 μM) in parotid-gland ductal cells obtained from wild-type or spinophilin-deficient mice to determine the role of this scaffolding protein in the regulatory actions of RGS2 protein on GPCR. It has been found that intracellular Ca2+ signaling in oocytes is regulated by RGS2 protein bound to spinophilin that associates with the third intracellular loop of α-adrenergic receptor [33]. Further, in neuronal dendritic spines and synapses, spinophilin is largely enriched and regulates glutamate transmission that is thought to be a key player for regulation of sympathetic outflow and autonomic control [32]. Spinophilin-deficient mice exhibit elevated blood pressure due to decreased parasympathetic activity [32] and a potentiated Ang II-mediated increase in blood pressure/heart rate [31]. As systemic knockout of spinophilin appears to cause disorders in the cardiovascular system, spinophilin may play a crucial role in vascular function. Nevertheless, there are no previous studies demonstrating function of the scaffolding protein in vasomotor responses. It will, therefore, be necessary to address the hypothesis in future studies as to whether the scaffolding protein, spinophilin, regulate activity of RGS proteins fine-tuning GPCR signaling that is crucial in cardiovascular system.
ROLES OF RGS PROTEINS IN THE CARDIOVASCULAR SYSTEM
There has been an exponential increase in studies aimed at understanding the functional significance of RGS proteins in the cardiovascular system. To clarify the physiological relevance of RGS proteins in vivo, genetic manipulation approaches such as knockout or knockdown of specific RGS proteins have been implemented (Table 2). For example, RGS2 protein deficient mice have shown promise as models for investigation of the physiological function of RGS2 protein. Thus, genetic ablation of RGS2 protein led to cardiac hypertrophy following prolonged Gαq stimulation (i.e. increased pressure overload evoked by transverse aortic constriction surgery), suggesting that mechanical stress mediates GPCR activation and subsequent cardiac hypertrophy in an RGS2 protein-dependent manner [34]. Earlier studies by Heximer and colleagues [35] led to the hypothesis that GPCR-mediated signaling contributing to blood pressure homeostasis utilizes precisely controlled negative feedback regulatory mechanisms that involve RGS2 protein. Mice either heterozygous or exhibiting a full knockout of RGS2 protein exhibit significantly increased mean arterial pressure (MAP) and purinergic receptor (P2Y)-mediated intracellular Ca2+ levels compared to controls [35]. RGS2 protein has also been shown to regulate nitric oxide (NO)-mediated vasodilation [36]. NO activates cGMP-dependent protein kinase (PKG) and subsequently leads to phosphorylation at the N-terminal of RGS2 protein that results in translocation toward plasma membrane and increased GAP activity of RGS2 protein [36].
In the heart and central nervous system RGS4 protein is the main species expressed and the transcriptional regulation of RGS4 protein appears to be altered in various pathophysiological situations. Interestingly and in contrast to other RGS proteins, pulmonary hypertension or increased pressure overload (by aortic band operation) upregulates mRNA expression for RGS4 protein in ventricular hypertrophy compared to control cardiac tissues [37-39]. Similar observations have been reported in human cardiac tissues where mRNA levels for RGS4 protein are 2-3 fold higher in cardiomyopathy compared to those in normal heart [40,41]. Therefore, it is likely that RGS4 protein acts as a compensatory regulator to prevent the heart failure from further progression. This is also supported by previous studies showing that Gαq-mediated hypertrophic signaling (i.e. myofilament arrangement and cardiomyocyte growth), in response to pressure overload, is reduced by overexpression of RGS4 protein [42].
Of the more than 30 RGS proteins, small arteries or arterioles display a strong expression of RGS5 protein [43]. Further, gene expression studies have identified that cardiovascular pathology is related to expression of RGS5 protein. In atherosclerotic lesions, mRNA levels for RGS5 protein are markedly reduced in arteriolar myocytes [44] while treatment with the anti-atherosclerotic agent, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor (statin), significantly upregulates expression of RGS5 protein in the aorta [45]. Moreover, transcription of RGS5 protein in the brain capillary and choroid plexus is markedly decreased in stroke-prone hypertensive rats [46]. Even though RGS5 protein is dispensable for arterial development during embryonic stages [14], the protein is required after birth for full arterial development [47,48]. Collectively the available data suggest that RGS5 protein may contribute to regulation of contractility or tone of arteries. Providing further support, gene expression studies show RGS5 protein to be a potent modulator of cardiovascular function. Knockout of RGS5 protein in arteriolar myocytes significantly potentiates Ang II-induced downstream signaling pathways, including enhanced activation of MAPK [21]. In cardiac tissue, pressure overload-induced cardiac remodeling is exacerbated in RGS5-deficient mice [49]. Further, in animal models with genetic deletion of RGS5 protein, resistance arteries are hypertrophied and markedly stiffened [8]. In RGS5 knockout mice, enhanced Ang II-mediated Ca2+ signaling and the resultant increase in femoral artery contraction are evident and may contribute to hypertension [8]. Aging-dependent vascular stiffness is further exacerbated in RGS5-deficient arteries [8]. In recent studies of pregnant RGS5-deficient mice, decreased expression of RGS5 protein has been implicated in causing gestational hypertension that is associated with enhanced contraction of femoral and uterine vascular beds in response to Ang II [50]. The RGS5 knockout-induced preeclampsia and gestational hypertension are ameliorated by an AT1R blocker (i.e. candesartan), suggesting that AT1R signaling-mediated blood pressure homeostasis is regulated by RGS5 protein [50].
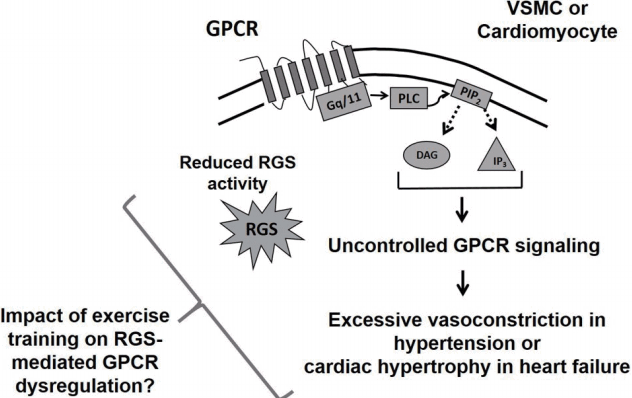
The above studies provide information supporting a protective role for RGS proteins in the cardiovascular system. In regard to this, upregulation of RGS5 protein has been observed to attenuate Ca2+ signaling and contractility of arteries in response to administration of GαqPCR-related vasoconstrictors, suggesting that Gαq protein-mediated downstream signaling pathways are modulated by RGS5 protein [8,21,38,51]. According to previous studies [49], RGS5 overexpression opposes cardiac hypertrophy and fibrosis caused by aortic banding (i.e. increased pressure overload/mechanical stress on cardiac myocytes), which result from the RGS5 protein-dependent inhibition of MEK-ERK1/2 signaling. Additionally, cardiac-specific RGS4 protein overexpression ameliorates Gαq protein signaling-dependent left ventricular hypertrophy under mechanical stress [52,53]. PKG-mediated phosphorylation of RGS4 protein, via application of atrial natriuretic peptide, plays an important role in guanylyl cyclase A-evoked attenuation of cardiac hypertrophy [37]. In that study, upregulation of RGS4 protein significantly abrogated heart weight, cardiomyocyte size, and cardiac hypertrophy-related gene expression in the guanylyl cyclase-A knockout animal model [37]. Taken together, it is suggested that RGS proteins mediate rapid and precise termination of GPCR signaling pathways and reduced RGS activity causes pathophysiological function of the cardiovascular system (Fig. 3).
EXERCISE TRAINING ON RGS PROTEINS
Preeclampsia is a prevalent cardiovascular risk factor and clinical disorder which shows proteinuria and hypertension after 20 weeks of gestation [54] and affects 2-8% of all pregnancies [55]. However, causes of preeclampsia remain incompletely understood. According to previous novel observations [50], it has been shown that expression of RGS5 protein in human myometrial arteries is significantly lower in hypertensive/preeclamptic pregnancies compared with normal pregnancies. In addition, in pregnant mice, RGS5 deletion have been shown to potentiate vascular contractility in response to Ang II and result in gestational hypertension [50]. Interestingly, it has been reported that the G allele of a polymorphism in RGS2 protein gene (i.e., C114G polymorphism in rs4606) is significantly related to decreased expression of RGS2 protein and hypertension/preeclampsia [56,57]. Women with the rs4606 CG or GG genotype in spiral arteries show a higher risk for hypertension during pregnancy [58]. This study also investigated a relationship between the gene polymorphism/hypertension and exercise and suggested that regular exercise intervention may reduce the prevalence of hypertension even in women who have the rs4606 CG or GG genotype [58].
Since previous studies investigating impact of exercise training on B/R4 subfamily RGS proteins are lacking, G protein-coupled receptor kinase (GRK) that is a different subfamily of mammalian RGS proteins would be introduced in this section. GRK2 has been shown to terminate GPCR signaling by desensitizing the receptor [59]. Recent studies have demonstrated that skeletal muscle-specific GRK2 knockout substantially attenuates the force of contraction of the extensor digitorum longus muscle [60]. This genetic animal model enhanced β2-adrenergic receptor (β2-AR)-mediated muscle hypertrophy by augmenting β2-AR/Akt-mediated pro-hypertrophic signaling, thereby suggesting that GRK2-dependent GPCR regulation is significant for skeletal muscle function and diseases [60]. Further, desensitization of β-AR by elevated GRK2 expression has been reported in hypertension or congestive heart failure [61,62]. These pathological disorders may result from a reduction in β-AR signaling-mediated vasodilation evoked by the increased GRK2 expression [63]. In contrast, free-floating swimming program (60 min per day, 5 days per week, 10 weeks) markedly decreased blood pressure and improved insulin sensitivity in spontaneously hypertensive rats (SHR) along with reductions in GRK2 expression and activity [64]. The ameliorated insulin sensitivity shown in swimming-exercised SHR led to a significant increase in vasodilation by enhancing insulin-Akt-endothelial nitric oxide (eNOS) signaling [64]. This convincing evidence suggests that exercise-induced alteration in GRK proteins may prevent the development or progression of cardiovascular diseases.
On the basis of GRK protein studies, it is conceivable that exercise training also increases or decreases expression at transcriptional or translational level, trafficking, or activity of RGS proteins within the cardiovascular system (Fig. 4). For instance, since RGS2 and 5 proteins regulate the signaling of endothelin, angiotensin, P2Y, or S1P receptors in VSMCs [11], it is likely that exercise training program may diminish those receptors-mediated excessive vasoconstriction or VSMC hypertrophy, critical hallmarks in hypertension, by increasing RGS2 and 5 protein expression and/or activity. However, while RGS protein-related modulation of GPCR signaling has been well-explored, the role of exercise training in RGS protein expression and activity remains to be investigated in detail. Hence, in the field of exercise science, the impacts of exercise intervention on cardiovascular RGS proteins and GPCR downstream signaling represent fertile areas for research (Figs. 3, 4).
SUMMARY
It is well established that GPCRs play a major role in signal transduction within the cardiovascular system. Further, dysregulation of GPCR-mediated downstream signaling likely leads to exaggerated or attenuated biological signaling responses that are directly associated with cardiovascular diseases. Therefore, the duration and/or magnitude of GPCR signaling is tightly controlled to sustain appropriate physiological functions in the cardiovascular system. As outlined earlier, RGS proteins hydrolyze GTP on the active α subunit of G protein and fine-tune G protein-mediated signaling. Thus, RGS proteins play an important role as negative feedback regulators and impaired RGS protein regulation has been strongly related to cardiovascular diseases. As exercise training has been demonstrated to reduce risk for cardiovascular disorders, it is therefore suggested that it will be valuable to explore the contribution of exercise training to RGS protein-mediated regulation of GPCR signaling within the cardiovascular system.