8주간의 저항성운동 및 열량 감소식이가 비만 중년 흰쥐의 Traf2-NFkB-mTOR 및 SIRT1-FoxO1 신호인자의 발현에 미치는 영향
Effects of 8 Weeks Calorie Reduction and Resistance Exercise on Traf2-NFkB-mTOR and SIRT1-FoxO1 Signal Expression of Cardiac Muscle in High-fat Induced Obese Middle-Aged Rats
Article information
Trans Abstract
PURPOSE
The purpose of this study was to investigate the effect of 8-week calorie reduction and resistance exercise on the expression of Traf2-NFkB-mTOR and SIRT1-FoxO1 signal of cardiac muscle in obese middle aged rats.
METHODS
50 weeks old male Wistar rats were induced to obese by high fat diet treatment for 6 weeks after 1 week of environmental adaptation and then were randomly assigned to 4 groups (control group, CON; diet control with calorie reduction group, DC; resistance exercise group, RE; diet control and resistance exercise group, DRE). After that ladder climbing exercise or calorie reduction with general formula were treated for 8 weeks.
RESULTS
Body weight and abdominal fat were significantly (p<.05) lower in the two groups as DC and DRE than the other groups as CON and RE. There was no significant difference in the weight of heart and skeletal muscle. Eight weeks of exercise or caloric reduction diet significantly (p<.05) reduced gene expression and activity of Traf2-NFkB-mTOR, which is a hypertrophic factor of cardiac muscle in obese middle-aged rats, and that autophagy factors of SIRT1-FoxO1 gene expression and activity were significantly (p<.05) increased. However, there was no synergistic effect with the combined treatment of the two stimuli.
CONCLUSIONS
These results suggest that caloric reduction with weight loss or resistance training may be effective in preventing cardiac hypertrophy and heart failure in middle-aged obese rats.
서 론
비만은 심장근육세포의 크기와 단백질 합성을 증가시켜 심장비대를 초래하며, 이는 심장 기능에 부정적인 영향을 미쳐 심장질환을 발생하게 한다[1-4]. 비만은 다양한 방법으로 심장의 형태와 기능의 이상을 초래하게 되는데, 심장의 지방산 산화의 변화[5], 심장세포의 산소소비량 증가[6], 산화스트레스 증가[7], 심장세포의 Ca2+ 조절 이상[8], 인슐린저항성 증가[9], 염증성 사이토카인 증가[10-12] 등이 있다. 그러나 비만과 심장비대의 인과관계를 증명할 수 있는 기전 규명은 여전히 미흡하다.
Tumor necrosis factor receptor-associated factor 2 (Traf2)는 면역과 염증반응, 세포사멸(apoptosis)과 같은 다양한 생물학적 활성을 조절하는 단백질로서 그 과발현은 유방암 세포 내 nuclear factor kappa-lightchain-enhancer of activated B cells (NFκB) 핵 전사를 활성화시켜 세포사멸을 억제한다[13,14]. 이러한 Traf2는 심장에서 높게 발현되고, 심장비대의 핵심요소인 NFκB 신호 전달에서 중요한 신호 단백질로서의 역할을 담당하는 것으로 보고되었다[15,16]. Divakaran et al. [16]은 심장에 제한적으로 Traf2가 과발현되는 유전자조작 생쥐(MHC-Traf2HC)를 대상으로 노화에 따른 심장근의 변화를 분석한 결과, 생쥐가 노화함에 따라 심근 내 전체 섬유소 콜라겐 함량이 증가하였으며 심장근육세포의 크기와 심장의 무게가 증가된 것을 확인하였다. 또한 Traf2는 phosphatidyl inositol3-kinase (PI3K)-protein kinase B (PKB) 신호전달기전을 활성화시키고 이는 glycogen synthase kinase-3β (GSK3β)의 불활성화 및 mammalian target of rapamycin (mTOR)의 활성화를 유도함으로써 궁극적으로 심근비대를 유도하게 된다[17].
Akt/mTOR 경로의 활성화는 심장세포의 자식작용(autophagy)을 저해한다[18]. 식이유도 비만과 대사증후군 모델에서 mTORC1 활성의 조절부전이 심장의 자식작용을 억제하게 되는데, Sciarretta et al. [19]은 비만 생쥐의 심장에서 mTORC1의 활성으로 자식작용이 유의하게 억제되고, 이는 심근의 허혈성 손상을 증가시키는 것으로 보고하였다. 심장세포에서 일어나는 자식작용은 심장의 정상적인 발달과 항상성 유지에 중요한 역할을 하는데[20,21], Silent information regulator-1 (SIRT-1)이 microtubule-associated proteins 1A/1B light chain 3B (LC3)와 autophagy-related protein 12 (Atg12)와 같은 자식작용과 관련된 유전자의 전사를 조절하는 FoxO1의 활성화를 증가시켜 조절하는 것으로 보고되었다[22-24].
인구의 고령화 및 비만, 당뇨병 발병률 증가 등으로 향후 심장마비 발생률은 증가할 것으로 예상된다[25]. 심부전 환자 수의 증가와 함께 전문적인 치료전략의 개발 및 적용에 투입될 비용은 국가와 개인 모두의 경제적 부담이 될 것이다. 현재 심부전 치료법은 없으며 심부전으로 인한 장기생존율은 매우 낮다. 환자의 3분의 1은 진단 1년 이내에 사망한다[26,27]. 따라서 다수의 연구가 심장비대 및 심부전을 예방하거나 역전시키기 위한 새로운 치료표적을 확인하기 위해서 심장비대 및 심부전으로의 전이와 관련된 분자기전을 규명하는 데 초점을 두고 있다.
지구성운동은 심장으로의 정맥회귀를 증가시켜 체적 과부하 및 편심비대(concentric hypertrophy)를 유발하지만[28,29], 근력운동은 체적 부하 및 동심 비대보다는 심장에 압력부하를 유발한다[28,30]. 또한 Barboza et al. [31]은 수컷 Wistar rats을 대상으로 유산소 및 저항성운동이 심근경색 이전에 훈련된 흰쥐의 심실 형태 계측 및 기능, 신체능력, 자율기능뿐만 아니라 심실 염증상태에 미치는 영향을 평가했다. 연구 결과 유산소운동과 같이 동적 저항성운동도 좌심실의 pro-inflammatory cytokine의 증가를 예방할 뿐만 아니라 심장과 혈관에 대한 교감신경의 영향을 감소시킨 것으로 나타났다. 즉 유산소운동뿐만 아니라 저항성운동에서도 유의한 심근보호효과가 나타남을 알 수 있다. 이상의 선행연구 결과들을 종합해 비만을 통한 혈압의 증가와 같은 압력 부하현상에 따른 심장의 병리학적 변화와 저항성운동을 통한 생리학적 변화를 분석하는 것은 운동프로그램의 처치효과를 제시하기 위한 중요한 근거를 제시할 수 있을 것으로 기대된다.
이에 본 연구는 비만에 의해서 초래된 심장비대에 따른 심장기능이상을 예방하고 처치하기 위한 방법을 모색함으로써 그 처치약물 개발에 도움을 주고자, 식이요법과 운동의 효과를 분자생물학적 관점에서 분석하고자 한다. 특히 심장의 생리학적 비대 원인인 운동의 효과를 기존의 지구성운동이 아닌 저항성운동을 통해 규명해보고자 한다.
연구 방법
1. 연구 대상
50주령 male Wistar rat 40마리를 대상으로 1주간 예비사육 후 6주간 고지방식 처치로 비만을 유도한 다음 4집단으로 무선 배정하여 8주간 실험을 실시하였다. 집단구성은 비교집단(control group; CON, n=10), 식이조절집단(Diet control [DC] group, n=10), 저항성운동집단(Resistance exercise [RE] group, n =10), 식이 조절+저항성운동집단(DRE, n=10) 등으로 한 케이지에 2마리씩 넣어 사육하였다. 사육실의 온도는 21°C로 유지하였으며 명기와 암기는 각각 12시간으로 조절하였다. 본 연구는 (재)대구테크노파크 바이오헬스융합센터 동물실험윤리위원회의 승인(BHCC-IACUC-2015-09)을 받아 실시되었다.
2. 연구절차
1) 식이 방법
고지방식이는 총 열량에 대해 carbohydrate 35%, fat 45%, protein 24%가 되도록 하였다(#D12451, Research diets, Inc). 식이조절집단은 고지방식에서 일반식(carbohydrate 64.5%, fat 11.8%, protein 23.5%, #D10001, Reserch diets, Inc)을 제공하는 방식을 사용하였다. 고지방식의 총열량은 4.73 kcal/g이었고, 일반식의 총열량은 3.0 kcal/g으로 제공하였다[32]. 전 실험기간 동안 사료와 물은 자유롭게 섭취하도록 하였다.
2) 저항성운동방법
저항성운동은 Jung et al. [32]이 제시한 사다리등반 운동프로그램을 실시하였다. 흰쥐들은 1주간의 사육실 적응 후 1주간 사다리등반운동에 적응하기 위하여 2일간은 꼬리에 무게를 달지 않고 연습하고, 하루 쉬고 2일간은 꼬리에 체중의 30-50% 정도 되는 무게를 달고 사다리등 반적응 훈련을 하였다. 6주간의 비만유도를 마친 후 운동그룹은 1 m의 긴 사다리에서 등반운동을 실시하였다. 사다리의 경사도는 80°이며, 꼬리에 무게를 달고 등반운동을 실시하였다. 사다리등반운동은 2분 간격으로 8회 반복하였다. 1세트에서는 체중의 70% 무게로 저항을 주고, 2-3세트는 체중의 80%, 4-5세트는 체중의 90%, 6세트는 체중의 100%, 7세트는 체중에 20 g을 더하고, 8세트는 체중에 40 g을 더하여 사다리등반운동을 실시하였다. 8주간 저항성운동은 주당 3회(월, 수, 금) 실시하였다. 운동 중간의 휴식기에는 사료와 물의 섭취를 자유롭게 하였다.
3) 조직 적출
Last-bout exercise effect를 배제하기 위해 8주간의 훈련 후 48시간의 회복기를 거친 다음 12시간 금식을 실시하였다. 70 mg/kg의 ketamine (JW Pharmaceutical, Korea)과 10 mg/kg의 Xylazine (JW Pharmaceutical, Korea)을 이용하여 마취하고 조직(muscle & heart)을 적출하였다. 적출된 근육들은 무게를 측정한 후 clamp frozen하여 분석 전까지 -80°C에서 보관하였다. 심장은 PBS로 관류하여 혈액을 완전히 제거한 후 적출하여 물기를 제거하고 무게를 측정한 후 좌심실을 분리하여 압착 동결하여 분석 전까지 -80°C에서 보관하였다. 복강 내 지방량을 측정하기 위해 epididymal, mesenteric, retroperitoneal fat pad를 적출하고 무게를 합산하여 산출하였다[33].
4) 측정항목 및 분석방법
(1) 체중 및 식이 섭취량 측정
실험기간 동안 체중과 식이섭취량은 격일로 오전 9시경 저울(Mettler PJ6, German)을 이용하여 소수 첫째 자리까지 측정하였다.
(2) Western blotting
적출된 심장은 ice-cold buffer (250 mM sucrose, 10 mM HEPES/1 mM EDTA [pH7.4], 1 mM Pefabloc [Roche], 1 mM EDTA, 1 mM NaF, 1 g/mL aprotinin, 1 g/mL leupeptin, 1 g/mL pepstatin, 0.1 mM bpV [phen], 2 mg/mL glycerophosphate 함유)로 균질화하였다. 균질화된 시료는 동결/해동 과정을 3회 반복하고 원심분리하였다(700 g, 10분). 원심분리된 상층액은 Lowry et al. [34]의 방법으로 단백질 정량을 하였다. 시료는 Laemmli buffer로 용해시켜 SDS-polyacrylamide gel에 분주한 다음 전기영동을 실시하였다. Immunoblotting 시 사용된 항체는 다음과 같다: Traf2 (Santa Cruz), SIRT-1 (Millipore), phospho-Akt, Akt, mTOR, phospho-FoxO1, FoxO1, NFkB, β-actin (Santa Cruz). 1차 항체처리 후 각각 2차 항체를 처리하고 ECL을 이용해 시각화한 다음 densitometry (sigma-plot 8.0 system)로 정량하였다.
3. 자료처리방법
측정된 모든 자료는 SPSS-PC (Version 21.0)를 이용하여 집단별 평균 및 표준편차를 산출하고 종속변인에 대한 집단 간 차이는 일원변량분석(one-way analysis of variance, ANOVA)을 이용하였다. 사후검정은 Duncan의 방법을 실시하였으며, 유의수준은 p<.05로 설정하였다.
연구결과
1. 체중의 변화
50주령의 중년 비만 흰쥐는 실험 환경적응 후 고지방식이를 이용하여 6주간 비만유도를 실시하였으며, 이후 무선배정 방식을 통해 4집단으로 나누어 8주간 식이 또는 운동을 실시하였다. 8주간의 처치기간 동안 고지방식이와 일반식이 집단의 1일 섭취열량은 유의하게(p <.05) 차이가 나타났으며, 운동처지에 따른 차이는 나타나지 않았다(Fig. 1A). 체중은 DC 집단과 DRE 집단이 CON 집단에 비해 처치 2주 후부터, RE 집단에 비해서는 3주 이후부터 통계적으로 유의하게(p <.05) 체중이 감소하였다(Fig. 1B).
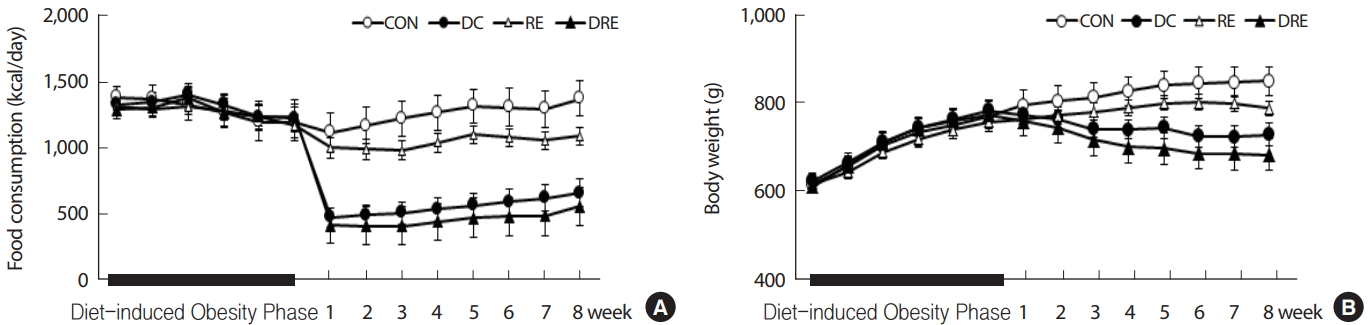
Change of (A) daily calorie intake and (B) body weight.
2. 근육량 및 심장, 복강 내 지방량의 변화
중년 비만 흰쥐를 대상으로 8주간의 식이 또는 운동처치 후 하지근육인 전경골근과 장지신근의 근육량은 집단 간 유의한 차이가 없었다(Fig. 2A, B). 또한 심장중량은 저항성운동집단이 다른 집단에 비해 높은 경향을 나타냈으나 집단 간 통계적 유의차는 나타나지 않았다(Fig. 2C). 그러나 복강 내 지방량은 식이조절집단(DC, DRE)이 CON 집단에 비해 유의하게(p<.05) 감소하였으며, 식이와 운동병행집단(DRE)의 복강 내 지방량은 다른 3집단에 비해 유의하게(p <.05) 감소한 것으로 나타났다(Fig. 2D).
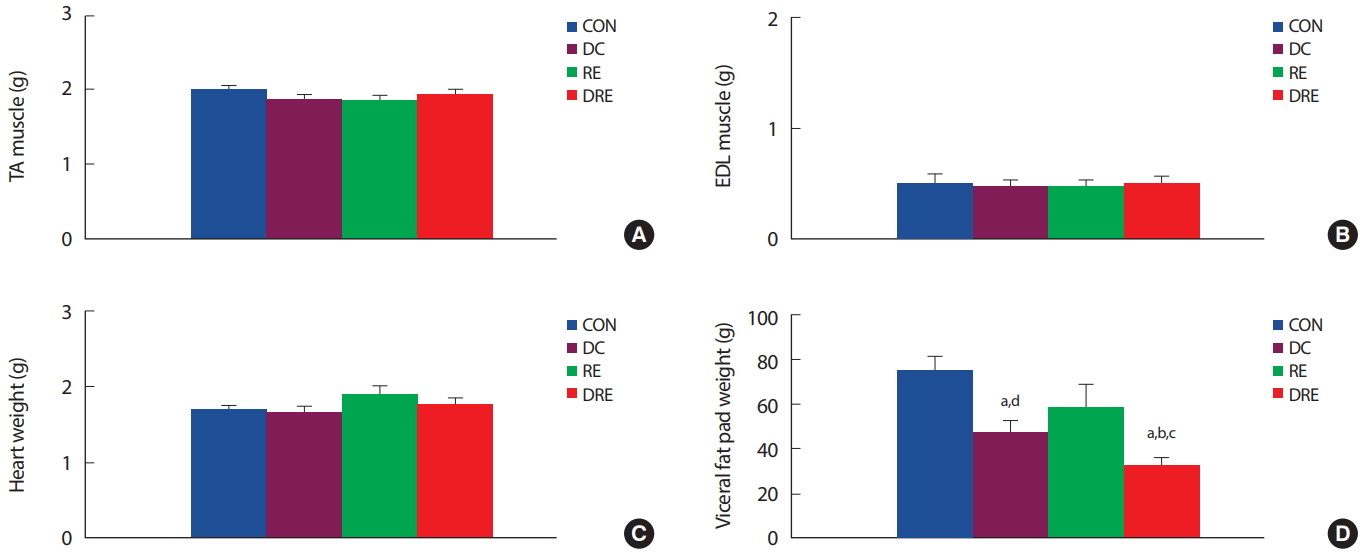
Differences in (A)Tibialis anterior, (B)Extensor digitorum longus, (C)Heart weight and (D) Abdominal fat mass. aSignificantly different from CON (p<.05); bSignificantly different from DC (p<.05); cSignificantly different from RE (p<.05); dSignificantly different from DRE (p <.05).
3. p-Akt/Akt protein expression ratio
중년 비만 흰쥐를 대상으로 8주간의 식이 및 저항성운동처치 후 심장근 내 유전자 발현의 변화를 알아보기 위해서 핵심 신호인자인 Akt의 활성도를 분석한 결과 4집단 간 유의한 차이가 나타나지 않았다(Fig. 3).
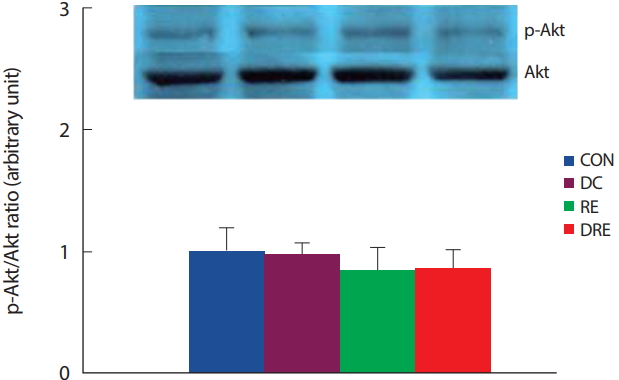
Differences in p-Akt/Akt ratio in cardiac muscle.
4. 심장근 비대관련 유전자 발현
8주간의 식이 및 저항성운동 후 심장근 내 심장근 비대관련 유전자(Traf2, NFkB, mTOR)의 단백질 발현의 변화를 분석한 결과, 식이 또는 저항성운동 모두 Traf2 단백질 발현이 유의하게(p <.05) 감소하였다(Fig. 4A). 또한 하위신호요인인 NFkB 발현도 식이 또는 운동으로 감소하였으며, 저항성운동집단이 비교집단에 비해 통계적으로 유의하게(p <.05) 감소하였다(Fig. 4B). mTOR 단백질 발현량은 식이 또는 저항성운동집단 모두에서 유의하게(p <.05) 감소하였다. 특히 식이조절 집단에서 mTOR 단백질 발현량이 가장 큰 폭으로 감소하였다(Fig. 4C).
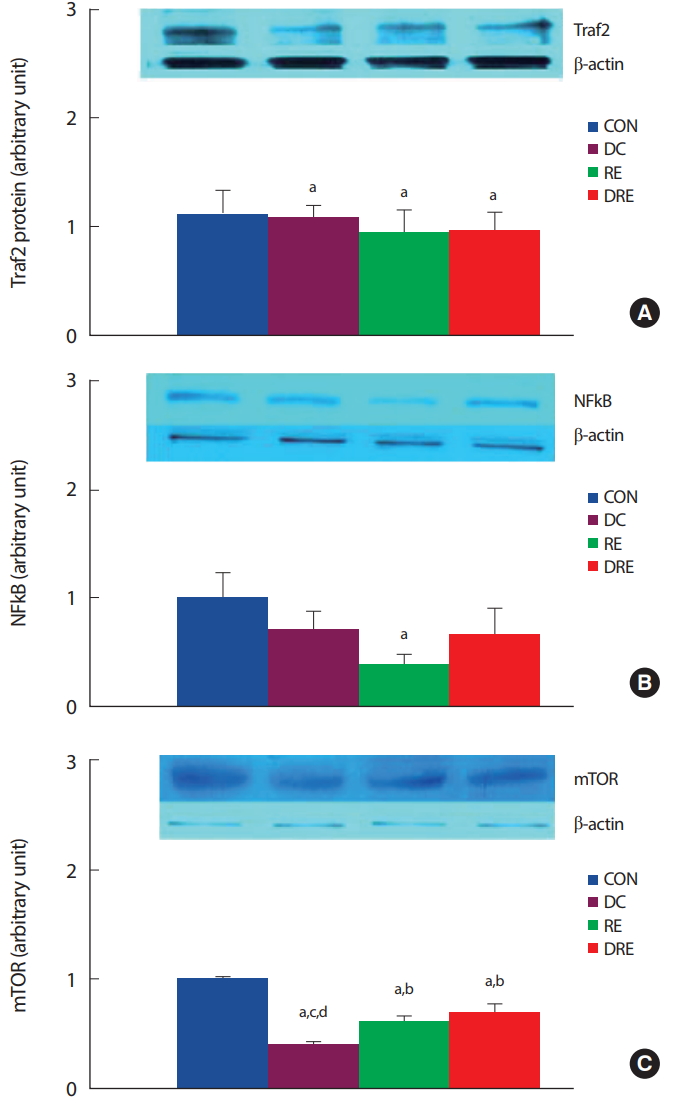
Differences in expression level of (A) Traf2, (B) NFkB and (C) mTOR in cardiac muscle. aSignificantly different from CON (p<.05); bSignificantly different from DC (p<.05); cSignificantly different from RE (p<.05); dSignificantly different from DRE (p<.05).
5. 심장근 내 자식작용 관련 유전자 발현
8주간의 식이 및 저항성운동 후 비만 중년 흰쥐의 심장근육 내 자식작용 관련 유전자(SIRT1, FoxO1)의 단백질 발현량을 분석한 결과, 식이와 운동처치 모두 심장근 내 SIRT1 단백질 발현을 유의하게(p<.05) 증가시켰으며, 저항성운동집단이 식이집단에 비해 유의하게(p<.05) 높게 나타났다(Fig. 5A). 하위신호인자인 FoxO1의 활성도는 FoxO1에 대한 phospho-FoxO1 단백질 발현비율로 산출한 결과, 식이 또는 저항성운동집단 모두 활성도를 유의하게(p<.05) 증가시켰다(Fig. 5B). 특히 저항성운동집단이 식이조절집단에 비해 FoxO1의 활성도를 유의하게(p <.05) 증가시켰으나 복합처치에 따른 부가적인 효과는 나타나지 않았다.

Differences in expression level of (A) SIRT1 and (B) p-FoxO1/FoxO1 ratio in cardiac muscle. aSignificantly different from CON (p<.05); bSignificantly different from DC (p<.05); cSignificantly different from RE (p<.05); dSignificantly different from DRE (p<.05).
논 의
비만은 현대인들의 주요 건강문제이며, 그 중요성은 끊임없이 증가하고 있다. 세계보건기구에 따르면 10억 이상의 성인이 과체중이며, 임상적으로는 적어도 3억 명 이상이 비만한 것으로 보고하였다. 비만은 심혈관 질환의 중요 위험인자이며, 허리-엉덩이 둘레 비율의 증가는 심근경색 발생률의 증가와 밀접한 관련이 있다[35]. 심장 보호전략을 연구하는 대부분의 동물연구는 젊고 건강한 동물에서 수행되었지만 비만과 같은 병적 상태가 심근보호능력을 저해한다는 것은 널리 알려져 있다[36-38]. 과도한 영양섭취와 비만에 의한 심근비대는 심부전 발생 시 선행되는 공통적인 조기 증상으로 수많은 신호전달경로에 의해 조절된다. Huang et al. [39]은 심근비대의 Traf2 역할과 관련하여 심장 특이적 Traf2 과발현 흰쥐의 심근세포변화를 분석한 결과, 심장에서의 Traf2 과발현은 심근을 현저하게 비대시켰으며, 좌심실 기능장애를 일으킨 것으로 보고하였다. 이와 같은 결과는 Traf2가 Akt/GSK3β 신호전달을 활성화시켜 심근비대를 강화시킨 것으로 보고하였다[40,41]. 활성화된 Akt는 다시 mTOR/S6K1 신호전달기전을 활성화시켜 내피세포의 노화를 초래하고 말초 허혈을 증가시킴으로써 심근비대증을 발생시킨다[42,43]. 본 연구의 결과에서도 중년의 식이유도 비만 흰쥐의 심장근 내 Traf2-NFkB-mTOR 발현이 저항성운동 또는 열량감소에 따른 체중감소 후 유의하게(p<.05) 감소하였다. 저항성운동의 경우 비교집단과 유의한 체중 차이가 나타나지 않았음에도 불구하고 심근비대신호인자의 감소를 나타냄으로써 선행연구의 결과와 일치하는 것으로 간주되며, 이와 관련하여 운동은 비만이나 과체중 환자의 심장사고에 따른 사망률을 감소시킬 수 있는 것으로 보고되었다[44,45]. Golbidi et al. [46]은 운동을 통한 심장보호 메커니즘을 크게 두 가지 범주로 나누어 설명하였는데, 활성산소종 생산 감소와 세포손상을 복구하는 메커니즘으로 나눌 것을 제안했다. 심장 내 mTOR는 Ying-Yang1-(YY1-) PGC-1α 경로를 통해 미토콘드리아 생합성 및 산화대사를 촉진하여 산화스트레스의 중요한 조절인자로 간주된다[47]. 또한 mTOR은 자식작용을 조절하고 미토콘드리아 제거를 증가시키며 PKB의 활성화를 통해 산화스트레스에 의해 유도되는 독성으로부터 심근세포를 보호한다[48-50]. 한편, 산화스트레스는 mTORC1을 조절하는데, 활성산소종의 생산은 GSK-3β 및 mTOR 신호전달을 저해한다[51]. 이와 같이 산화스트레스와 mTOR 신호사이의 관계는 mTOR가 산화스트레스를 조절할 뿐만 아니라 활성산소종의 활성화에 mTOR 신호체계가 영향을 받기 때문에 매우 복잡하다. 본 연구에서는 손상복구기전에 대한 접근만 시도되었으며, 활성산소종에 대한 분석은 실시되지 않았다. 따라서 추후 보다 세부적인 분석을 통해서 분명한 규명이 이루어져야 할 것으로 생각된다.
본 연구에서는 자극에 따른 심근 내 비대요인의 발현뿐만 아니라 자식작용 관련요인도 분석함으로써 운동자극과 열량 제한에 따른 체중감소가 심장근의 항상성에 미치는 영향을 비교 분석하였다. 분석 결과 심근 내 자식작용을 일으키는 주된 신호인자인 SIRT1-FoxO1 신호전달 체계의 발현 및 활성도가 운동 또는 식이제한 후 유의하게 증가하였다. 또한 전술한 것처럼 mTOR의 발현은 유의하게 감소하였다. 본 연구의 결과와 유사하게 Qu et al. [52]은 심근 내 PARP1-SIRT1-mTOR 신호전달 체계는 심장질환 및 당뇨병 발생과 높은 관련성이 있는 것으로 보고하였으며, Xie et al. [53]은 심근 내 자식작용은 mTOR 신호전달계에 의해서 영향을 받는데, mTORC1의 발현을 억제하면 자식작용이 증가하는 것으로 나타났다[54,55]. 본 연구에서 한 가지 흥미로운 결과는 저항성운동과 식이조절의 복합처치에 따른 상승효과가 나타나지 않았다는 것이다.
본 연구의 결과만으로 정의하기 어려우나 저항성운동 또는 식이섭취열량 감소를 통한 체중감소가 중년 비만 흰쥐의 심근 내 유사한 신호전달기전을 통해서 비대/자식작용을 나타낼 것으로 생각된다. 그러나 추후 연구를 통해서 더욱 분명하게 규명될 필요가 있다. 본 연구에서는 선행연구와 다르게 처치에 따른 Akt 활성의 변화가 나타나지 않았다. Akt는 serine/threonine-specific protein kinase로 활성도가 오래 지속되지 않는 특성이 있다[56]. 따라서 본 연구에서 실시된 조직 채취시점이 운동 자극 후 48시간 이후였던 점을 감안한다면 분석시점과 활성시점이 일치하지 않았기 때문으로 생각된다.
결 론
8주간 저항성운동 또는 식이섭취열량의 감소를 통한 체중 감량이 비만 중년 흰쥐의 심근 내 근비대 및 자식작용 관련 유전자들에 미치는 영향을 분석한 결과, 저항성운동 또는 열량감소식이는 심근 내 비대요인의 발현/활성은 저하시키고 자식작용 관련 유전자들의 발현/활성은 증가시켰다. 그러나 두 가지 자극의 복합처치에 따른 상승작용은 나타나지 않았다. 이러한 결과를 통해서 체중감소를 동반하는 열량 조절식이 또는 저항성운동트레이닝은 중년 비만 흰쥐의 심장비대 및 심부전을 예방하는 데 효과적인 것으로 생각된다.