유산소 운동이 제2형 당뇨 쥐 해마에서 미토콘드리아 칼슘 조절과 세포사멸에 미치는 영향
Effects of Aerobic Exercise Training on Mitochondrial Ca2+ Homeostasis and Apoptosis in the Hippocampus of Type 2 Diabetic Rats
Article information

Abstract
Abstracts
PURPOSE
Type 2 diabetes mellitus (T2DM) is a recognized risk factor for neurodegenerative diseases such as Alzheimer's disease (AD). Mitochondrial dysfunction caused by disturbance of calcium (Ca2+) homeostasis might be a pathophysiological link between T2DM and AD. In the present study, we investigated the effects of aerobic training on the regulation of mitochondrial calcium homeo-stasis, mitochondrial function (O2 respiration), and apoptosis in the hippocampus of T2DM rats.
METHODS
Thirty male Otsuka Long-Evans Tokushima Fatty rats were divided into three groups: control group (CON, n=10), diabetes control group (DM, n=10), and diabetes+aerobic exercise group (DM+EXE, n=10). The rats in the exercise group were forced to run on a treadmill for 30 min, three times a week for 8 weeks.
RESULTS
The DM group showed significantly increased mitochondrial calcium uniporter (MCU) protein and apoptosis-related factors such as Bax and caspase-3 and decreased mitochondrial O2 respiration when compared with the CON group. However, the hippocampus of rats in the DM+EXE group exhibited a significant decrease in MCU, Bax, and caspase-3 levels and an increase in Bcl-2 and mitochondrial O2 respiration when compared with the DM group.
CONCLUSIONS
These results suggest that regular aerobic exercise alleviates mitochondrial dysfunction and apoptosis by modulating MCU, which is involved in mitochondrial Ca2+ homeostasis in the early stages of T2DM. This might be a potent strategy to prevent neurodegenerative diseases.
서 론
제2형 당뇨병(type 2 diabetes mellitus, T2DM)의 주요 특징인 인슐린 저항성과 만성적 고혈당은 중추 신경계 영역의 손상을 유도함으로써 경도 인지 장애, 알츠하이머 병(Alzheimer's disease, AD) 및 모든 유형 의 치매를 포함한 신경퇴행성질환의 발생 위험을 증가시킨다[1,2]. 최근 T2DM 환자에서 인지 기능 저하 및 AD 유병률의 지속적인 증가[3] 추세와 더불어 다양한 신경퇴행성질환의 발생에 대한 주요 위험 인자로 T2DM이 제안됨에 따라 이들 사이의 상호관련성을 규명하기 위한 역학(epidemiological), 인지 및 신경 병리학적 수준에서의 활발한 연구가 진행되고 있다[4].
특히, 중추신경계에서 미토콘드리아 기능장애는 T2DM과 신경퇴행성질환 사이의 병태생리를 이해하기 위한 핵심적 요인으로 제안되고 있다[5]. 미토콘드리아는 신경세포의 생존과 흥분에 필요한 에너지 공급, 이온 항상성 및 세포 사멸의 시작을 포함한 핵심적인 과정에 관여한다[6]. 반면 특정 요인에 매개된 미토콘드리아 이상(mitochondrial abnormalities) 및 기능장애는 인지장애 관련 질환 발생과 밀접한 관련성을 지닌다. 이와 관련된 선행연구에서, T2DM에 매개된 미토콘드리아 기능 및 신호전달 장애는 신경퇴행성질환의 주요 병리적 특징인 세포사멸을 초래함으로써 신경세포 손실을 유도하는 것으로 보고된 바 있으며, 이와 더불어 신경퇴행성질환에서 관찰되는 ATP 생성 감소, ROS 생성 증가 및 투과성 전이 기공(permeability transition porel, PTP) opening은 미토콘드리아 기능장애에 따른 결과로 간주된다[7,8].
이러한 미토콘드리아 기능장애 발생은 세포 내 칼슘(Ca2+) 이온 농도 및 항상성에 의존적이다. Ca2+은 세포 운명(cell fate)을 좌우하는 핵심적인 요소로, 미토콘드리아 내 Ca2+ 수준은 신경 활성 및 ATP 생성과 같은 일련의 생물학적 과정에서 중요한 역할을 담당한다[9]. 세포질 및 미토콘드리아에서 과도한 Ca2+ 축적은 미토콘드리아 기능장애 발생에 대한 주요 원인일 뿐만 아니라 과도한 ROS 생성을 초래함으로써 programmed cell death 및 기타 생물학적 신호전달을 유발하는 것으로 보고되었다[10]. 이와 관련된 선행연구를 살펴보면, 미토콘드리아 내 Ca2+ 항상성 교란은 미토콘드리아 생합성 및 에너지 대사를 저해함으로써 시냅스의 손상을 초래하고[11], 특히, T2DM 상태에서 중추신경계 ROS 생성 및 신경세포사멸은 세포질 및 미토콘드리아 Ca2+ 유입의 조절 실패 및 Ca2+ 통로의 기능적 결함에 기인된 결과로 보고된 바 있다[12]. 또한 Ca2+ 과부하는 세포 내 손상을 초래하는 주요 자극으로써 미토콘드리아 세포사멸 경로를 촉진시키는 것으로 보고된다[13]. 이에 비추어 볼 때, 미토콘드리아로의 Ca2+ 이온 유입 조절은 T2DM에서 신경퇴행성질환 발생으로 이어지는 병리적 과정을 보다 더 잘 이해하기 위한 필수적 요인임을 유추해 볼 수 있다.
미토콘드리아로의 Ca2+ 이온 유입 및 방출은 원형질막, 미토콘드리아 및 소포체에 위치한 선택적 복합체(mitochondrial calcium uniport-er, MCU)를 통해 조절된다[14]. MCU는 뇌를 포함한 다양한 조직에서 미토콘드리아 내막에 위치한 Ca2+ 유입의 주요 통로로서[15], MCU에 매개된 정상적인 Ca2+ 유입은 다양한 기질 효소의 활성화에 의한 metabolisms-secretion coupling을 용이하게 하고 산화적 인산화에 의한 ATP 생성을 촉진시킨다[16]. 반면, MCU 기능적 결함 및 과발현은 미토콘드리아 내 Ca2+ 수준을 교란시킴으로써 신경퇴행의 초기 단계에서 발견되는 세포사멸을 촉진시키는 것으로 보고되었다[17]. 이처럼, MCU에 매개된 Ca2+ 항상성 교란은 신경퇴행성 질환 발생에 관여하는 핵심적인 세포 내 변화이며[18], 또한 미토콘드리아 Ca2+ 항상성의 이상 조절(dysregulation)이 T2DM 상태의 다양한 조직에서 관찰됨을[19] 감안할 때, 세포 및 미토콘드리아 내 Ca2+ 농도에 관여하는 MCU 발현 조절은 T2DM 상태에서 향후 신경퇴행성 질환의 예방 및 개선을 위한 중요한 요인이라 할 수 있다. 비록 이전 선행연구에서, 미토콘드리아 기능장애 및 흥분독성에 의한 신경세포사멸 예방을 위한 새로운 치료적 전략으로서 MCU 발현 조절의 중요성이[20] 제안된 바 있지만 아직까지 MCU 발현 조절에 기여하는 효과적인 중재 전략 탐색에 대한 연구는 국내외적으로 희소한 실정이다.
운동은 T2DM에 유도된 중추 영역의 손상을 완화하고 신경퇴행성질환 발생의 예방을 위한 효과적인 비약리적 전략으로 잘 알려져 있다. 운동은 뇌 가소성, 인지 기능 개선에 관여하고 노년기 인지 능력 저하 및 치매 위험을 감소시키는데 효과적인 전략 중 하나로 보고된다[21]. 특히, T2DM으로부터 유도되는 만성적 스트레스에 가장 취약한 영역인 해마에서 분자적·대사적 변화는 신경퇴행성 질환 발병의 주요 기전으로 보고된다[22]. 반면, 운동에 매개된 해마 조직 산화스트레스 및 염증 반응의 억제는 기억력의 개선 및 향상에 긍정적 영향을 미치는 것으로 보고된다[23,24]. 추가적으로 T2DM과 신경퇴행성 질환 발생 사이의 공유된 인자로 밝혀진 해마 영역의 신경전달 장애 및 β-amy-loid 축적 등에 대한 운동 중재 효과는 다수의 선행연구를 통해 입증되고 있다[25,26]. 최근 미토콘드리아가 T2DM과 신경퇴행성 질환 발생에 관여하는 또다른 중요 요인으로 제안되고 있지만, 아직까지 T2DM 에서 후기 신경퇴행성 질환 발생에 이르는 병리적 과정에서 미토콘드리아의 실제적 기능, Ca2+ 항상성 조절 인자 및 세포사멸에 대한 운동 효과 연구는 미비한 실정이다. 이에 본 연구에서는 초기 T2DM 단계에서 규칙적인 운동 중재가 해마 조직 내 미토콘드리아 산소 소비량, MCU 발현 및 세포사멸에 미치는 영향을 규명하고자 하였다.
연구 방법
1. 연구 대상
제2형 당뇨 표현형을 가진 (Otsuka Long-Evans Tokushima Fatty, OLETF) 모델 쥐(n=20)와 대조군 LETO 모델 쥐(n=10) 30마리를 무작위 표본 추출법을 이용하여 당뇨 대조군(CON, n=10), 당뇨군(DM, n=10), 당뇨+ 운동군(DM+EXE, n=10)으로 구분하였다. 최초 공급받은 쥐를 온도 22-24°C, 습도 60±5%가 유지되며, 밤낮주기가 자동 조절 장치에 의해 조절되는 사육실에서 연구 목적에 부합되는 주령(16-21 weeks)까지 철저한 관리를 통해 사육하였다. 식이는 실험기간 동안 고형사료(단백질 22.5%, 지방 3.5%, 저섬유 7.5%, 회분 9.0%, 칼슘 0.7%, 인 0.5%)와 물을 충분히 섭취하도록 하였다. 동물실험 윤리위원회의 승인(HNU 2018-01)을 받았으며, 실험 수행동안 동물의 고통과 불편을 최소화하도록 노력하였다.
2. 운동 프로토콜
동물용 소형 트레드밀 기기를 이용하여 총 8주 동안 유산소 운동을 실시하였다. 본 운동에 앞서 모든 실험동물의 스트레스 방지와 적응을 위해 1주간의 트레드밀 적응 훈련을 10 m/min 속도로 15분간 실시하였다. 이후, 운동강도 및 빈도 설정은 선행연구를[27] 참고하여 주 3회, 1일 30분간 20 m/min 속도로 운동을 실시하였다.
3. 조직 시료 준비
실험 종료 후 12시간 이상 공복상태를 유지한 후, Ketamine (50 mg/ mL)과 2% Xylazin (23.3 mg/mL)을 3:1로 혼합하여 마취한 후 해마 조직을 적출하였다. 해마 조직 일부(3-5 μ g)를 적출 후 즉시 미토콘드리아 기능 분석을 위해 Oroboros Oxygraph-2K Systerm 장비에 넣고 분석하였으며, 나머지 조직은 증류수를 이용하여 혈액을 제거하고 dry ice-isopentane 용액으로 급속 동결시킨 후 –80°C에 보관하였다.
4. Western Blot
조직을 protease와 phosphatase inhibit cocktail이 첨가된 lysis buffer (50 mM HEPES, 10 mM EDTA, 100 mM NaF, 50 mM sodium pyro-phosphate, 10 mM sodium orthovanadate, 1% Triton at pH 7.4)로 ho-mogenization 하고 4°C, 14,000 rpm에서 15분 동안 원심분리 하였다. 상층액을 BCA Protein Assay Kit을 이용하여 단백질 농도를 측정한 후, 30 μ g의 단백질을 8-10%의 SDS-polyacrylamide gel에 전기영동 시켰다. 이후, gel상의 단백질을 100 V에서 1시간 동안 nitrocellulose membrane 에 transfer 하였다. Membrane을 5% non-fat milk에서 1시간 동안 blocking 처리하고 TBST buffer로 세척한 후 1:1,000으로 희석한 1차 항체를 overnight 하였다. 이후 TBST 용액으로 10분씩 3차례 세척한 후 2차 항체 Anti-rabbit IgR를 0.5% BSA/TBS를 사용하여 1:3,000으로 희석시킨 용액에 membrane를 넣고 1시간 동안 shacking 시킨 후 항원과 결합하지 않은 2차 항체를 TBS용액으로 10분간 3회 세척하였다. 마지막 단계로 ECL detection reagent를 이용하여 Hyperfilm에 현상하고 스캔한 후, 이미지 분석 프로그램 image J를 이용하여 산출하였다.
5. 미토콘드리아 산소 소비량
미토콘드리아 기능을 분석하기 위한 요인으로, Oroboros Oxygraph-2K Systerm (Innsbruck, Austria)을 이용하여 미토콘드리아 산소 소비량을 측정하였다. 약 3-5 μ g의 해마 조직을 buffer [130 mM sucrose, 60 mM C6H11O7K, 1 mM EGTA, 3 mM MgCl2, 10 mM K2HPO4, 20 mM HEPES와 1 mg/mL BSA(pH 7.4)]에 넣고 다음과 같은 프로토콜을 이용하여 미토콘드리아 산소 소비량을 측정하였다. (i) 2 mM glutamate+ 1 mM malate (complex I substrate), (ii) 2 mM ADP (state 3 condition), (iii) 3 mM succinate (complex II substrate). 미토콘드리아의 O2 소비량은 picomoles/sec/mg tissue weight로 나타내었다.
6. 자료처리
측정된 결과 분석은 Graph pad Prism 8.0 프로그램(GraphPad Soft-ware, Inc., La Jolla, CA, USA)을 이용하여 각 항목별 평균 및 표준편차를 산출하였다. 각 변인들의 그룹간 차이 검증은 일원분산분석(one-way analysis of variance, ANOVA)을 이용하였으며, 사후검정은 Tukey's multiple comparison test를 실시하였다. 이때 유의수준은 .05로 설정하였다.
연구 결과
1. 운동에 의한 MCU 단백질 변화
8주간 운동 중재에 따른 해마 조직 내 MCU 발현을 비교 분석한 결과는 Fig. 1과 같이 집단 간 통계적으로 유의한 차이가 나타났다(p =.001). DM 집단은 CON 집단과 비교하여 MCU 단백질 수준이 통계적으로 유의하게 증가된 것으로 나타났다(p <.001). 반면, 트레드밀 운동을 실시한 DM+EXE 집단은 DM 집단과 비교하여 통계적으로 감소된 것으로 나타나(p <.05), 규칙적인 유산소 운동이 미토콘드리아 Ca2+ 채널에 긍정적 변화를 가져올 수 있음을 확인할 수 있었다.
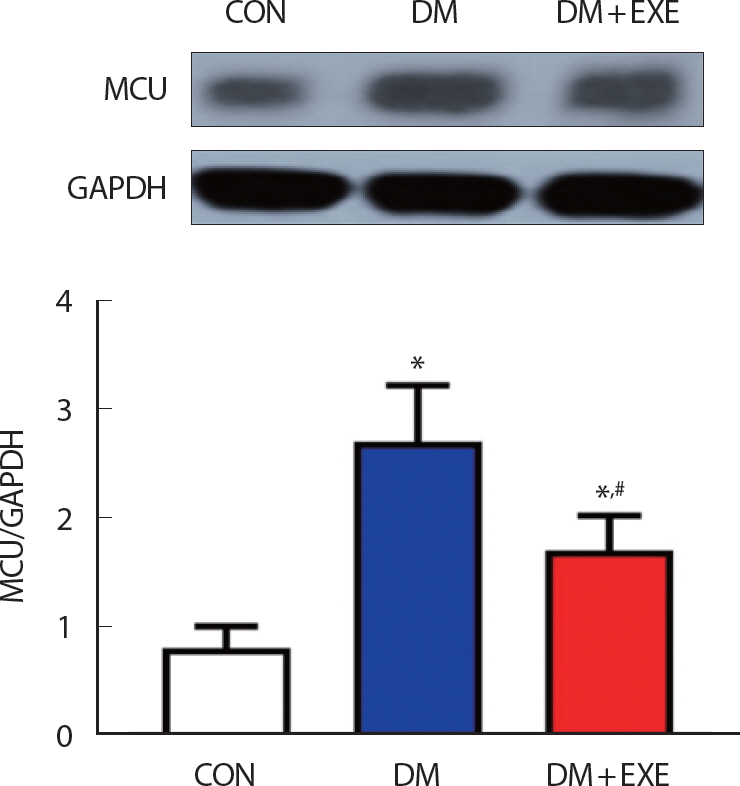
Comparisons of MCU protein expression. All data are presented as the Mean±SD. Significantly different from CON, * p<.05; Significantly dif-ferent from DM, # p<.05.
2. 운동에 따른 미토콘드리아 O2 소비량 변화
해마 조직 내 미토콘드리아 산소 소비량을 비교 분석한 결과는 Fig. 2와 같이 집단 간 유의한 차이가 나타났다(p =.001). 미토콘드리아 com-plex I (glutamate+malate, GM), State 3 condition (ADP) 및 complex II (succinate, SUCC)의 모든 기질 하에서 DM 집단은 CON 집단과 비교하여 통계적으로 낮은 산소 소비량을 나타내었다(p <.05). 반면, 트레드밀 운동을 실시한 DM+EXE 집단은 DM 집단과 비교하여 통계적으로 유의하게 증가된 산소 소비량(p <.05)을 보임으로써, 유산소 운동이 T2DM에 매개된 미토콘드리아 산소 소비 감소를 개선시킴을 확인할 수 있었다.
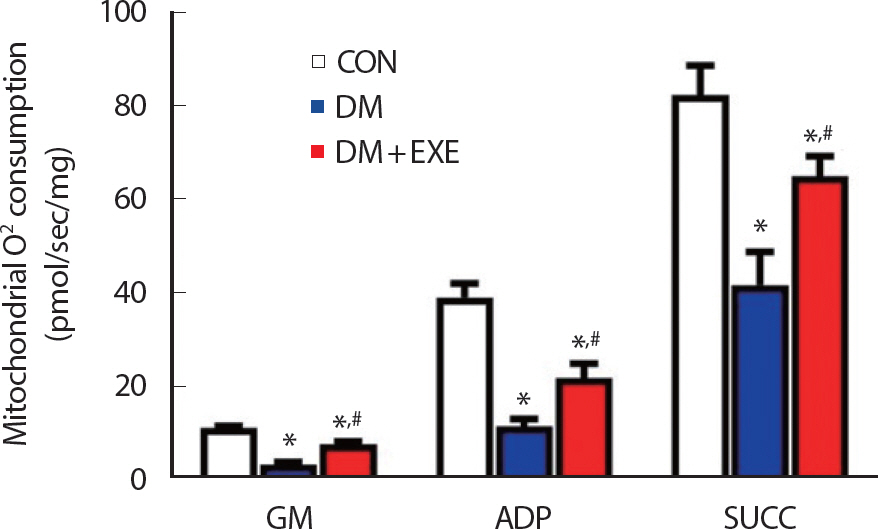
Comparisons of mitochondrial O2 consumption. All data are pre-sented as the Mean±SD. Significantly different from CON, * p<.05; Signifi-cantly different from DM, # p<.05.
3. 운동에 따른 세포사멸 변화
8주간 운동 중재에 따른 세포사멸 관련 인자에 대한 비교 분석 결과는 Fig. 3과 같이 집단 간 통계적으로 유의한 차이가 나타났다(p = .001). 사후 검증 결과, DM 집단은 CON 집단과 비교하여 Bax 및 caspase-3 단백질 수준이 통계적으로 유의하게 증가된 것으로 나타났다(p <.001, p <.05). 반면, 트레드밀 운동을 실시한 DM+EXE 집단은 DM 집단과 비교하여 통계적으로 감소된 것으로 나타났다(p <.05). 또한, Bcl-2 단백질 수준에서 DM 집단은 CON 집단과 비교하여 통계적으로 유의하게 낮게 나타났으며, DM+EXE 집단은 DM 집단에 비해 통계적으로 유의하게 높게 나타났다. 이와 더불어, Bcl-2/Bax ratio는 DM 집단이 CON 집단과 비교하여 통계적으로 유의하게 감소(p <.05)된 반면, DM+EXE 집단은 DM 집단과 비교하여 통계적으로 유의하게 증가(p <.01)되는 것으로 나타났다.

Comparisons of apoptosis-related factors. All data are presented as the Mean±SD. Significantly different from CON, * p<.05; Significantly different from DM, # p<.05.
논 의
T2DM의 유병 기간이 길어질수록 신경퇴행성질환의 발생 위험은 증가하고, T2DM 상태에서 중추 영역의 분자적 · 대사적 변화 및 신호전달 장애는 신경퇴행성질환에서 관찰되는 양상과 유사한 수준을 보인다[28]. 특히, 중추신경계의 생리적 과정에 핵심적 요인으로 알려진 Ca2+의 이상조절은 미토콘드리아 기능장애를 유도하고 다양한 뇌 영역의 손상에 관여한다. 따라서, 미토콘드리아와 Ca2+ 이온 사이의 관련성은 T2DM을 포함한 만성적 대사 장애와 신경퇴행성 질환 사이의 병태생리를 보다 더 잘 이해하기 위한 중요한 요인일 지 모른다. 이에 본 연구는 중추신경계에서 미토콘드리아 Ca2+ 항상성 조절 인자에 대한 운동 매개 효과를 규명한 최초의 연구라 생각되며, 연구 결과를 통해 T2DM이 해마 영역 내 미토콘드리아 Ca2+ 통로 MCU의 과발현을 유도 함으로써 미토콘드리아 기능 저하 및 세포사멸 경로 활성에 관여함을 확인하였다. 이와는 대조적으로 T2DM 초기부터 규칙적인 운동에 의한 MCU 발현 억제, 증가된 미토콘드리아 산소 소비량 및 항-신경세포사멸 효과를 확인할 수 있었다.
미토콘드리아로의 Ca2+ 유입은 에너지 생산에서부터 세포사멸에 이르기까지 다양한 세포 항상성 과정에 기여할 뿐만 아니라 신경망의 신경 기능과 구조적 적응을 조절한다[29]. MCU는 미토콘드리아로의 Ca2+ 유입과 유출을 조절함으로써 세포 내 Ca2+ 수준의 패턴을 변화시키기 때문에, 미토콘드리아에 의한 Ca2+ 완충 능력은 Ca2+ 의존적 신호전달 조절 및 다양한 질병의 병태생리에 중요한 역할을 한다[30]. 선행연구에서 Ca2+ 이상조절에 따른 미토콘드리아 기능장애는 T2DM과 주요 신경퇴행성질환 모두에서 관찰됨으로, 신경세포 Ca2+ 신호 및 농도 수준 조절은 노화, T2DM 및 신경퇴행성 질환과 관련된 인지장애를 개선하기 위한 효과적 치료제 개발에 매우 중요한 요인으로 제안되고 있다[31]. 또한, 미토콘드리아 Ca2+ 농도 및 MCU 발현 조절이 AD와 신경흥분 독성 장애에 대한 치료 표적으로 보고된 바 있다[32]. 비록 중추 영역에서 미토콘드리아 Ca2+에 대한 운동 중재의 직접적인 영향을 규명한 선행 연구가 희소한 실정임으로 다각적인 논의는 제한적이지만, 중추 및 말초 영역에서 미토콘드리아 수, 크기, 형태 및 역동성은 운동에 의해 조절될 수 있으며[33], 규칙적인 운동이 전반적인 뇌 기능 개선과 함께 T2DM과 신경퇴행성 장애 사이의 주요 공유된 인자에 긍정적 변화를 유도할 수 있음은 또다른 선행연구에서 보고되고 있다[34]. 이에 비추어 볼 때, 운동은 미토콘드리아 막에 위치한 통합적 채널의 조절에도 영향을 미칠 수 있음을 짐작할 수 있다. 따라서, 본 연구에서 나타난 운동 매개 해마 조직 MCU 발현 억제는 미토콘드리아로의 Ca2+ 유입 속도 지연 및 농도 수준 감소에 영향을 미침으로써 Ca2+ 항상성 조절에 기여할 수 있을 것으로 사료된다.
추가적으로, 본 연구에서 MCU 과발현에 따른 미토콘드리아 com-plex I, II 기질 하에서 산소 소비량 감소 및 세포사멸 관련 인자의 유의한 증가가 DM 집단에서 나타났다. 이는 미토콘드리아 기능 및 세포사멸에서 MCU 발현 양상의 중추적인 역할을 시사하는 결과로 생각된다. 이와 관련된 선행연구에서 Ca2+ 과부하는 미토콘드리아 complex I 및 II와 같은 전자전달계 (electron transport chain, ETC)의 손상을 초래함으로써 포도당 대사를 교란시키고 ATP 생성 감소 및 ROS 생성을 증가시키는 것으로 보고되며[35], 미토콘드리아 complex I 기능 저하는 AD를 포함한 신경퇴행성 질환의 발병에 잠재적인 통합요소로 간주된다. 과인산화된 tau 단백질로 구성된 세포 내 신경섬유엉킴(neurofibril-lary tangle) 및 세포 외 Aβ plaques는 AD의 주요 특징이며, 과인산화 tau 단백질은 미토콘드리아 complex I 손상을 유도하여 ROS 수준을 증가시키고 ATP 생성을 감소시키는 것으로 보고되었다[36,37]. 또한, 뇌 인슐린 저항성에 유도된 미토콘드리아 산소소비 감소 및 대뇌 영역 에서의 포도당 대사율 변화는 T2DM과 관련된 인지 기능 저하의 주요 요인으로 보고된다[38].
이와 더불어, MCU 발현 증가에 따른 Ca2+ 과부하는 미토콘드리아에서 pro-apoptosis 관련 인자의 증가에 기여한다[39]. Ryan et al. [40]은 MCU 과발현에 따른 미토콘드리아로의 증가된 Ca2+ 유입은 산화적 인산화를 자극하여 ROS 생성을 증가시킴으로써 산화스트레스, mPTP 개방, 세포사멸 및 신경퇴화를 촉진하는 것으로 보고하였다. Liao et al. [41]은 MCU 과발현이 N-methyl-D-aspartate (NMDA) 매개된 Ca2+ 유입을 증가시켜 미토콘드리아 막전위 감소를 초래하는 반면에 MCU knockdown은 Ca2+ 수준을 감소시켜 막전위 감소 및 흥분독성 세포사멸을 예방할 수 있음을 보고하였다. 세포사멸 조절 유전자 발현 장애는 인슐린 저항성 및 만성적 고혈당으로 유도된 신경 세포사에 대한 주요 경로이다. 특히, 산화스트레스 증가, 항산화 매개 변수 감소, Bax 증가 및 Bcl-2 감소는 T2DM 모델 쥐의 주요 뇌 영역에서 관찰되는 대표적인 현상으로 보고된다[42]. T2DM에 의한 해마 손상과 관련하여, Kahya et al. [43]은 Ca2+ 유입 및 산화스트레스 과부하가 기억 및 학습에 관여하는 해마 영역의 손상을 초래하는 주요 원인임을 보고하였고, Xu et al. [44]의 연구에서는 db/db mice와 T2DM 환자의 사후(post-mortem) 해마 조직에서 caspase-3 및 Bax 단백질 발현이 증가되는 반면 Bcl-2 및 Bcl-2/Bax ratio는 감소되는 것으로 나타났다. 이 외에도 해마 조직에서 인슐린 저항성 및 만성적 고혈당에 기인된 Bax, Cyt-c, caspase-3의 유의한 증가는 다수의 선행연구에서 관찰되었다[45,46]. 반면, 미토콘드리아 기능 및 세포사멸 억제에 대한 운동 효과는 다양한 선행연구를 통해 입증되었다. 당뇨병에서 운동으로 인한 caspase-3, 8 활성 감소는 산화적 손상의 감소와 관련되며, 규칙적인 운동은 Bcl-2/Bax ratio을 증가시킴으로써 항-세포사멸 효과를 나타낸다[47]. Lu et al. [48]은 STZ (streptozotocin)에 유도된 AD 쥐의 해마에서 미토콘드리아 기능 장애 및 신경 세포 사멸은 운동에 의해 개선됨을 보고하였다. 이 연구에서 운동은 낮은 ATP 수준과 관련된 cytochrome c 산화 효소의 활성 및 ATP 생성을 향상시킴으로써 미토콘드리아 에너지 대사에 긍정적인 영향을 미칠 뿐만 아니라 caspase-3, −9 단백질 수준을 현저히 감소시키는 것으로 나타났다. 이러한 선행연구와 일치되는 경향으로, 본 연구에서도 T2DM 그룹에서 미토콘드리아 산소 소비량의 유의한 감소와 함께 세포사멸 인자 Bax, caspase-3 발현 수준이 증가된 반면 규칙적인 운동은 이러한 요인의 개선에 긍정적 영향을 미치는 것으로 나타났다.
결 론
종합해 보면, T2DM과 신경퇴행성질환 사이에서 중요한 병리적 특징인 미토콘드리아 기능 장애 및 세포사멸은 중추 영역의 MCU 발현 수준에 의존됨을 확인할 수 있었다. 반면, 운동은 MCU 발현을 감소시킴으로써 미토콘드리아 산소 소비량 증가 및 세포사멸 억제에 긍정적 영향을 미치는 것으로 나타났다. 비록 더 많은 연구가 필요하지만, T2DM 상태에서 중추신경계 미토콘드리아 Ca2+ 항상성 유지 및 개선을 위한 MCU 발현 조절은 향후 신경퇴행성질환의 발생을 예방하기 위한 새로운 잠재적 표적이 될 수 있으며, 운동은 이에 대한 치료적인 가치를 제공할 것으로 사료된다.
Notes
The authors declare that they do not have conflict of interest.
AUTHOR CONTRIBUTION
Conceptualization: SH Park; Data curation: SH Park; Formal analysis: SH Park; Funding acquisition: SH Park; Methodology: SH Park, JH Yoon; Project administration: SH Park, JH Yoon; Visualization: SH Park; Writing-original draft: SH Park; Writing-review & editing: JH Yoon.