서론
노화는 신체의 모든 조직에 영향을 미치는 여러 인자에 의해 생기는 과정이며 노화에 대한 연구는 퇴행성 신경질환이나 골질량 감소 등에 초점이 맞추어져 왔고 최근에 와서야 노화로 인한 근육감소증(sarcopenia)의 연구가 시작되었다. 근육감소증은 나이가 들면서 골격근의 근질량과 기능의 생리적 감소 현상을 말한다[1]. 점진적인 근육 질량의 손실은 노인들에게 건강 위험을 유발할 수 있는 데, 예를 들면 신체 활동을 감소시키고 넘어질 가능성을 증가시켜 골절을 일으킬 수 있다. 부상 후에도 재활 시간이 종종 지연되어 비활동성 위축(disuse atrophy)을 일으켜 침대에서의 장기요양의 결과를 낳는다[2]. 노화에 의한 근육감소증은 고령자들에 있어서 독립적인 생활과 관련있는 자립 생활 체력의 저하뿐만 아니라 당뇨병이나 심혈관질환과 같은 노화 관련 질환의 조기 유발과도 밀접한 관련이 있다[1]. 근육감소증의 병리학적 원인은 복잡하고 많은 요인들이 기여하는 것이 특징이며 증가된 세포사멸(apoptosis)이 근육감소증의 중요한 원인이라는 연구가 있어 왔다[3-5]. 근육감소증의 진전의 또 다른 중요한 요인은 단백질의 합성(protein synthesis)과 단백질의 분해(protein degradation)의 불균형이다[6]. 근육은 단백질 합성과 단백질 분해 사이의 균형, 즉 단백질 전환율(protein turnover)에 의해 근질량이 유지된다. 단백질 합성의 증가와/또는 단백질 분해의 감소는 정적인 단백질 균형(positive protein balance)으로 단백질이 축적된다. 반면, 부정적인 단백질 균형(negative protein balance)은 단백질 합성의 감소와/또는 단백질 분해의 증가로 인해 골격근의 단백질 손실이 생긴다. 노인들의 근육 손실은 단백질 합성률의 감소, 단백질 분해률의 증가, 또는 이 두 가지 과정의 조합이 원인이 될 수 있다[7]. 따라서 노화와 관련된 근육감소증은 단백질 전환율의 변화와 관련이 있는 듯하다. 인간을 대상으로 한 연구들에서 노화된 근육의 기초 단백질 분해는 변화가 없거나[8,9], 약간 증가하는 것으로 보고되었다[10]. 하지만 놀랍게도 기초 단백질 합성은 젊은 사람과 노인 사이에서 전혀 차이가 발견되지 않았다[10,11]. 따라서 노화된 골격근에서 근육 감소증은 단백질의 합성보다는 단백질의 분해와 더 깊이 관련되어 있는지 모르겠다.
골격근은 크게 2가지 단백질 분해 경로를 갖고 있다. (1) 자가포식-리소솜 분해 체계(the autophagy-lysosome system)와 (2) 유비퀴틴-프로테아솜 시스템(the ubiquitin-proteasome system, UPS). UPS가 골격근의 주요한 단백질 분해 경로로 여겨지고 있으며 골격근의 수축성 단백질을 분해하는 중요한 단백질 분해 체계라고 한다[6,12,13]. 젊은 쥐에서 단식, 비신체적 활동(physical inactivity)이나 질병(예; 암, 전신염증, 심부전증 등)에 의해 생긴 근 위축(muscle atrophy)이 일어나는 동안, 단백질의 유비퀴틴화(ubiquitination)와 UPS에 의한 분해가 현저히 증가한다고 알려져 있으며, 특히 근원섬유 단백질의 분해가 UPS에 의해 일어난다[12]. 반면, 자가포식-리소솜 체계(여기서부터 자가포식으로 부름)는 세포막 표면 단백질과 세포 밖의 엔도사이토시스(endocytosis)된 단백질을 분해하며[14], 생명이 긴(long-lived) 단백질과 세포소기관을 분해시키는 비선택적인(non-selective) 과정으로 알려져 있다. 이 두 분해 체계들은 골격근의 단백질 항상성(homeostasis) 유지에 중요한 역할을 하며 노화 과정 중에 변화가 있는 듯하다. 또한, 노화 관련 근육 감소증과 밀접하게 연관되어 있는 것으로 여겨진다. 운동과 같은 생리적인 자극이 노화된 골격근의 단백질 분해에 영향을 미치고 근육 감소를 지연시킬 수 있다는 제안이 있어왔지만 그러한 가능성에 대한 정확한 생리학적 기전은 아직 잘 알려져 있지 않으며 일치하지 않은 연구 결과 그리고 사실이 확립되어있지 않은 상태이다.
골격근과 같은 세포 분열이 끝난 세포(post-mitotic cell)에서 줄기세포로부터 새로운 세포가 대치되지 않기 때문에 항노화(anti-aging) 효과와 같은 세포 보호 기능은 단백질 전환(protein turnover)에 크게 의존하게 된다. 무척추 동물과 고등 생물에서 정상적인 노화 과정 중에 자가포식은 감소한다고 보고되었다[15,16]. 비효율적인 자가포식은 분해되지 않아 리소솜에 붙어있는 lipofusin (리소솜 내부의 분해되지 않은 복합체), 단백질 축적물(aggregates), 손상된 미토콘드리아와 같은 노화와 관련된 세포의 손상된 물질이 축적되는 주요한 원인이 된다[17]. 노화 쥐의 간을 분석한 결과 자가포식체와 리소솜의 결합 능력의 감소로 인해 자가포식에 의한 단백질 분해가 감소하였다[18]. 반면 노화 쥐에서 자가포식 활성이 증가되어 있다는 증거 또한 보고되었다. 젊은 쥐(3-4개월)와 노화 쥐(30-31개월)의 다양한 유전자 발현을 분석한 결과 노화 쥐의 가자미근에서 리소솜 내부의 단백질 분해 효소(protease) 인 cathepsin L의 유전 발현의 증가가 발견되었다[9]. 하지만 다른 연구(3개월과 30개월의 쥐의 비복근)에서는 이 결과를 확인시켜 주지 못하였다[19]. 자가포식체의 형성에 중요한 역할을 하는 Beclin-1이 노화 쥐의 plantaris 근육에서 유의하게 증가를 보였지만 Atg7과 Atg9의 단백질은 젊은 쥐와 비교해 차이가 없었다[20]. 젊은 쥐(3개월)에 비해 노화 쥐(22개월)의 대퇴근에서 LC3 I이 LC3 II로의 전환율(자자포식 유도를 나타내 주는 생화학적 지표)이 크게 증가하였다[21]. 나이와 관련해서 자가포식의 변화 정도는 조직에 따라 달라지는 것 같다. 노화와 함께 간에서 자가포식 활동은 감소하지만[16], 들쥐의 노화된 심장에서 자가포식은 그대로 유지된다고 제안되었다[22]. 현재까지 우리가 알고 있는 지식으로는, 노화와 함께 골격근에서 자가포식의 변화는 아직 명확히 정립되어 있지 않은 것이 사실인 듯하다.
따라서 본 연구는 노화된 근육에서 자가포식 유동을 조사하였고 지구력 운동훈련이 노화된 골격근의 자가포식 유동에 미치는 영향을 알아보는 데 목적을 둔다. 따라서 이 연구는 선행연구에서 설명된 것처럼[23,24], 자가포식 유동을 측정하여 자가포식의 활성을 조사하였다. 즉, 자가포식을 억제하는 약물인 colchicine을 사용하는 경우와 사용하지 않는 경우를 동시에 두고 autophagosome marker 단백질들인 LC3 II(mitrotubule-associated protein light chain 3)와 p62 (Sequestosome 1, SQSTM1)의 turnover assay를 사용하여 8주 수영훈련이 자가포식 유동에 미치는 영향을 조사하였고 자가포식과 관련된 단백질 수준을 측정하여 자가포식의 변화를 살펴보았다.
연구 방법
1. 실험동물
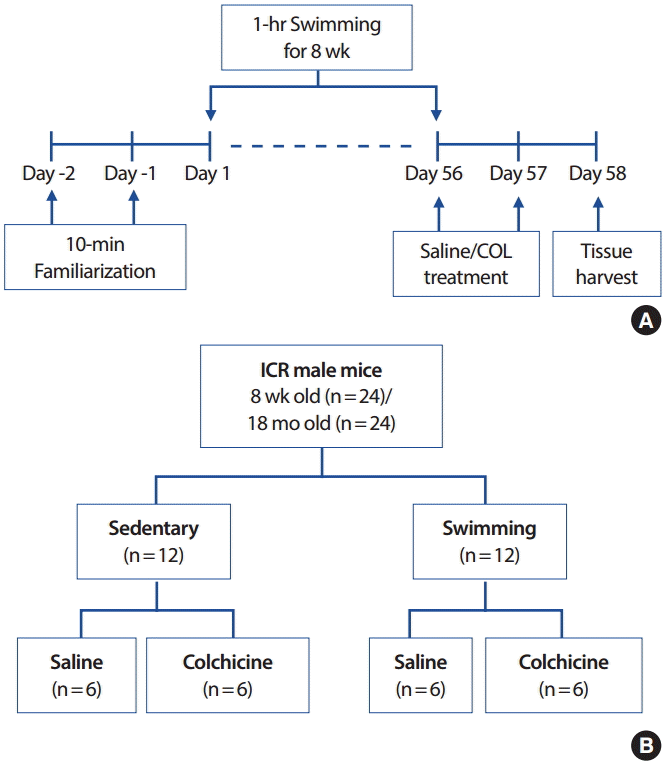
본 연구에서 사용된 실험동물은 생후 8주령 male ICR 마우스 24마리와 male 노화 쥐(18개월) ICR 마우스 24마리(샘타코)를 구입하여 1주간의 적응기를 마친 후, 각각 6마리씩 무작위로 임의 배정하여 실험에 사용하였다. 사육실의 온도는 22℃, 습도는 약 50%, 명암은 12시간 주기로 조절하였다. 사료와 물은 충분히 공급하고, 실험동물 취급법에 따라 실험하였으며, S 대학교 동물실험윤리위원회의 승인을 받아 실시하였다.
2. 실험방법
1) 운동 프로그램
Fig. 1A에서 설명된 것처럼, 수영 운동은 2일간의 10분 수영 적응을 마친 후 35-36℃의 물에서 1시간 한 차례 수행되었다. 이 수영 훈련은 1주일에 5일, 8주 동안 지속되었다. 물과 사료는 자유롭게 먹을 수 있게 되었으며, 22℃의 온도와 50%의 습도로 설정된 동물 채임버에서 실험이 끝날 때까지 사육되었다.
2) In vivo autophagy flux assay
지구성 운동이 마우스 골격근의 자가포식 유동을 변화시키는 가를 조사하기 위해, in vitro autophagy flux assay를 동물모델에 적용시켜 개발된 “in vivo autophagometer” [23] 방법을 사용하였다. 미세소관 중합억제제(microtubule depolymerizing agent)인 colchicine (Col, 0.4 mg/kg/day, Sigma-Aldrich, #C9754)을 처치하는 그룹과 처치하지 않는 그룹을 포함시켜 LC3 II를 Western blot으로 측정하였다. Fig. 1B에 설명된 것처럼, 골격근에서 in vivo autophagy의 변화를 측정하기 위해 쥐를 희생시키기 전 이틀 동안 colchicine i.p. 주사 그룹과 saline i.p. 주사 그룹을 포함시킨 4개의 그룹을 적용시켰다(control+saline, control +colchicine, exercise+saline, exercise+colchicine).
3) Western blotting 분석
마우스 골격근에서 자가포식 유동의 측정은 전기영동법(Westrn blot)에 의해서 분석되었다. 이 실험에서는 Bio-Rad사의 Western blot 시스템을 사용하여 전형적인 형태의 전기영동법을 사용하여 특정한 단백질의 양을 분석하였다. 간단히 설명하면, 상완삼두근(triceps muscle) 을 그라인더에 protease inhibitors cocktail (Sigma-aldrich, #P2714)이 섞인 ripa buffer 안에서 분쇄되어 lysates로 만들어졌다. BCA assay를 통해 전체 단백질 양이 조사된 뒤 SDS와 염색약과 함께 섞어 샘플을 준비하였다. 단백질은 전기영동에 의해 젤에서 분리되고 nitrocellulose membrane (0.2 μm, Bio-rad)에 전이시킨 후 5%의 우유에 blocking을 하였다. 그 후 primary항체와 함께 overnight 4℃에서 incubation을 시키고 washing을 실시한 뒤 secondary 항체로 incubation을 시켰다. 다시 washing과정을 거친 뒤 ECL 용액(Pierce Biotechnology)에 incubation되고 필름에 현상되어 특정 단백질 수준을 분석하였다. Band의 강도는 densitometric scanning을 하여 “ImageJ” (NIH) 프로그램을 통해 단백질 양이 수량화(quantification)되었다. 이 실험에서 사용된 Primary 항체는 anti-LC3B (L7543), anti-actin (A2066), Sigma-Aldrich; anti-p62 (1842-1-AP), Proteintech; anti-Beclin-1 (3738), anti-Bnip3 (13795), anti-HDAC6 (D21B10), Cell Signaling Technology; anti-Lamp1 (MABC39), Millipore이다.
연구 결과
1. 체중과 음식 섭취량 변화
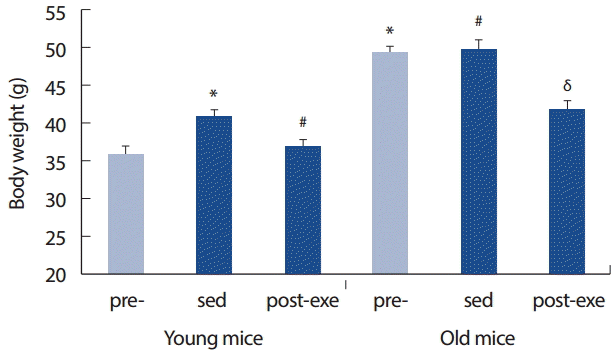
젊은 쥐의 비훈련 통제그룹(sedentary)은 8주 동안 유의한 몸무게의 증가(p <.05, -5 g)를 보였지만 수영훈련 그룹은 유의한 몸무게의 증가(-1 g)가 나타나지 않았다(Fig. 2). 반면 노화 쥐의 경우 비훈련 통제 그룹은 8주 동안 몸무게의 변화가 없었지만 8주 수영 훈련이 노화 쥐의 몸무게를 -7.5 g 감소시켰다(p <.05) (Fig. 2).
Table 1에서 보인 것처럼, 일일 식이섭취량에 있어 수영 훈련에 참가한 젊은 쥐는 비운동 통제그룹에 비해 섭취량이 더 많은 경향(△ -1.1 g)을 보였지만 노화 쥐는 훈련 그룹과 비훈련 간의 식이섭취량이 크게 나타나지 않았다(△ -0.2 g).
2. 노화가 마우스 골격근의 자가포식 유동에 미치는 영향
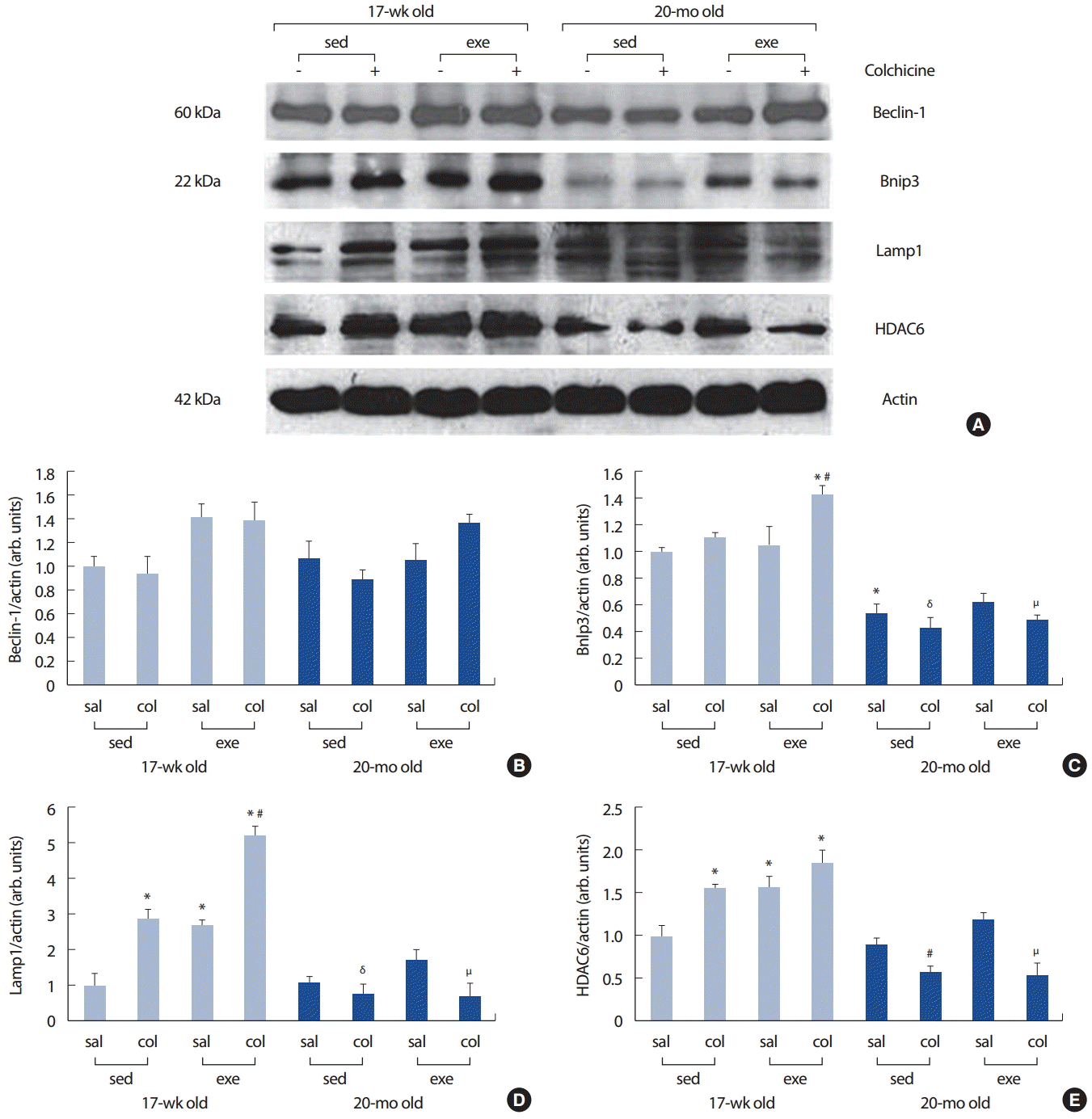
골격근의 자가포식이 노화에 의해 변화하는 가를 알아보기 위해, 젊은 쥐(17주)와 노화 쥐(20개월)의 골격근은 채취하여 Western blot 분석을 통해 자가포식 유동을 조사하였다. 생체 자가포식 유동 분석 결과 젊은 쥐는 saline이 처치된 골격근에 비해 colchicine이 처치된 골격근에서 LC3 II/actin이 -300%가 증가되었다(p <.05) (Fig. 3A, B). 반면 노화 쥐는 saline이 처치된 골격근에 비해 colchicine이 처치된 골격근에서 LC3 II/actin이 -200%가 증가되었다(p <.05) (Fig. 3A, B). 17주 젊은 쥐에 비해 20개월 노화 쥐의 골격근에서 자가포식의 유동이 -33%가 감소되었다. 또 다른 자가포식의 유동을 측정하는 방법인 LC3 II/LC3 I 전환율을 보면, 젊은 쥐는 saline이 처치된 골격근에 비해 colchicine이 처치된 골격근에서 -300%가 증가되었지만(p <.05) (Fig. 3A, C), 노화 쥐는 saline이 처치된 골격근에 비해 colchicine이 처치된 골격근에서 -100%가 증가되었다(p <.05) (Fig. 3A, C). LC3 단백질과 함께 흔히 사용되는 자가포식 지표인 p62는 자가포식이 유도될 때 감소하고 자가포식이 억제되면 증가한다[25]. Saline처치 그룹에서 젊은 쥐와 노화 쥐의 p62 단백질 양의 차이는 없지만 colchicine처치 때, 젊은 쥐에 비해 노화 쥐의 p62의 단백질 양이 -50% 증가하였다(Fig. 3A, G). 이는 노화와 함께 자가포식의 활성이 크게 감소하였음을 말해 준다.
3. 지구성 훈련이 노화 쥐의 골격근의 자가포식 유동에 미치는 영향
8주 지속적인 운동이 젊은 쥐와 노화 쥐의 골격근의 자가포식에 미치는 영향을 조사하기 위해, 8주의 수영 훈련을 시킨 뒤 in vivo autophagy flux assay 방법을 사용하여 노화 쥐와 젊은 쥐의 골격근의 자가포식 유동을 측정하였다(Fig. 4). 8주 수영 훈련이 젊은 쥐의 골격근에서 자가포식의 유동을 100% 증가시켰다(p <.05, sed+col vs. exe+col) (Fig. 3A, F). 반면 8주 수영훈련은 노화 쥐의 골격근에서 자가포식의 유동을 통계적으로 유의하게 변화시키지 못하였다(p <.05, sed+col vs. exe+col) (Fig. 3A, F). 또한 8주 수영 훈련이 젊은 쥐(17주)의 골격근의 LC3 II/LC3 I 전환율을 증가시킨 반면 노화 쥐(20개월)의 골격근에서 LC3 II/LC3 I 전환율을 변화시키지 못하였다(Fig. 3A, E). 이 결과는 골격근의 p62의 단백질 수준의 변화와도 일치한다. 젊은 쥐의 경우 p62의 단백질 수준이 운동과 colchicine을 처치했을 때 운동과 saline을 처치한 그룹에 비해 유의한 증가(-125%)를 보여주었지만(p <.05) 노화 쥐들은 두 그룹 간의 유의한 증가가 나타나지 않았다(Fig. 3A, E). 젊은 쥐에 비해 노화 쥐의 골격근은 8주 수영 훈련에 반응하여 자가포식 유동을 증가시키지 못하였다.
4. 지구성 훈련이 노화 쥐의 골격근의 자가포식과 관련된 단백질 발현에 미치는 영향
초기 자가포식체(autophagosome) 형성에 필수적인 단백질인 Beclin-1(Atg6)은 두 연령 간 그리고 모든 처치에 유의한 차이가 나타나지 않았다(Fig. 4A, B). 젊은 쥐의 골격근에서 8주 수영 훈련이 mitophagy receptor로 알려진 Bnips3 단백질 수준을 colchicine 처치와 함께 유의하게 증가시켰다. 하지만 노화 쥐의 골격근에서는 Bnip3 단백질이 젊은 쥐보다 감소되었으며 유산소 훈련에 대해 미미한 반응을 보여주었다(Fig. 5A, B). Bnip3처럼 노화 쥐의 골격근에서는 리소솜 지표인 lysosomal-associated membrane protein 1 (Lamp1) 단백질이 젊은 쥐보다 감소되었으며 8주 수영 훈련에 대한 반응을 나타내주지 않았다(Fig. 5A, C). Fig. 4A, D에서 보이는 것처럼, 8주 수영훈련이 젊은 쥐의 골격근의 자가포식 촉진 인자인 microtubule-associated deacytylase 6 (HDAC6) 단백질 수준을 유의하게 증가시킨 반면 노화 쥐에서는 HDAC6 단백질 수준이 운동 훈련에 의해 유의한 증가를 보이지 않았다. 또한 이틀간의 colchicine 처치가 노화 쥐의 HDAC6 단백질 수준을 유의하게 감소시켰다(Fig. 5A, D).
논의
본 연구는 노화된 골격근에서 단백질 분해 시스템 중의 하나인 자가포식 시스템의 유동(flux)을 조사하였고 지구성 운동 훈련이 노화에 의해 변화된 골격근의 자가포식의 유동에 간섭(intervention)을 일으키는 가를 조사하였다. 연구방법에서 설명된 것처럼 자가포식의 유동은 자가포식 억제제인 colchicine을 처치하는 그룹과 처치하지 않는 두 그룹을 동시에 두고 자가포식 지표인 LC3 II (가장 흔히 사용되는 자가포식 지표) 단백질의 변화를 통해 자가포식의 활성을 측정할 수 있는 생체 자가포식 유동 분석법(in vivo autophagic flux assay)을 사용하여 조사되었다.
본 연구에서 골격근의 자가포식의 유동이 노화에 의해 변화하는 가를 알아보기 위해, 젊은 쥐(17주)와 노화 쥐(20개월)의 골격근을 채취하여 전기영동법을 통해 자가포식의 유동을 측정하였다. 자가포식의 유동은 자가포식의 개시(initiation)와 해결(resolution)까지 이루어지는 모든 과정들을 포함한다[26]. 자가포식의 유동이 증가되었다는 것은 자가포식의 반응(response)뿐만 아니라 분해 능력(degradation capacity) 또한 증가된 것을 말한다. 생체 자가포식 유동 분석 결과, 젊은 쥐의 골격근에 비해 노화 쥐의 골격근에서 자가포식의 유동이 약 33%가 감소하였고, LC3 I이 LC3 II로 전환되는 비율(LC3 II/LC3 I) 또한 젊은 쥐의 골격근에 비해 노화 쥐의 골격근에서 자가포식의 유동이 약 55% 감소되었다(Fig. 3). LC3 II/LC3 I 전환율은 자가포식 유동을 측정하는 또 다른 방법으로 자가포식체(autophagosome)에 결합되지 않는 형태(nonlipidated form of LC3, LC3 I)가 자가포식체에 결합되어 있는 형태(phosphatidylethanolamine, PE-conjugated form, LC3 II)로 전환되는 비율(ratio)을 측정하는 방법으로 자가포식체의 합성(synthesis)의 정도를 나타내 준다. 이 측정 방법(자가포식 억제제를 사용하지 않고 LC3 II/LC3 I 전환율만을 측정)은 경우에 따라 자가포식의 유동을 분석하는데 잘못된 해석을 낳기도 한다[24]. 젊은 쥐와 노화 쥐의 colchicine 처치가 없는 그룹(두 sed+sal 그룹)을 비교해보면 노화 쥐의 LC3 II/actin 단백질 수준과 LC3 II/LC3 I 전환율이 약 200% 증가되었다. 많은 자가포식 연구자들이 이 결과를 자가포식이 노화 쥐에서 더 증가되었다고 해석을 한다. 하지만 이것은 젊은 쥐에 비해 노화 쥐의 골격근에서 자가포식이 증가되어서가 아니라 노화 쥐의 골격근에서 자가포식의 분해 능력이 감소되어 LC3 II가 근섬유 내에 더 많이 측적되었기 때문으로 해석하는 것이 맞다. 따라서 이 결과들을 종합해 보면 젊은 쥐에 비해 노화 쥐의 골격근에서 자가포식 유동이 유의하게 감소된 것을 알 수 있으며 생체 자가포식 유동 분석법이 자가포식의 활성화를 측정하는데 자가포식의 유동에 대한 잘못된 해석(misinterpretable)을 방지해줄 수 있는 아주 중요한 분석 방법이라는 것을 말해 준다. LC3 I만 살펴보면, 모든 그룹 간의 유의한 차이가 나타나지 않았다(Fig. 3A, D). 노화 쥐도 젊은 쥐처럼 자가포식의 시작(initiation), 즉 자가포식의 유도(induction)는 차이가 나타나지 않았지만 젊은 쥐와 달리 노화 쥐는 자가포식의 해결(resolution), 즉 분해시키는 능력은 유의하게 감소되어 있다는 것을 의미한다. 이는 노화 쥐는 젊은 쥐에 비해 골격근의 자가포식의 유동이 감소되어 있는 것을 뜻한다. 노화 쥐에서 자가포식의 개시(initiation)가 젊은 쥐처럼 손상되어 있지 않다는 것은 Beclin-1(Atg6)의 Western blot의 결과로 또한 알 수 있다(Fig. 3A, E). Beclin-1은 초기 자가포식체 형성에 필수적인 단백질로 본 연구의 젊은 쥐와 노화 쥐의 골격근에서 Beclin-1의 차이가 나타나지 않는 것으로 보아 초기 자가포식체의 형성에는 노화에 의한 손상이 없음을 알 수 있다. 본 연구에서 젊은 쥐와 노화 쥐의 triceps 근육에서 Beclin-1 단백질 수준의 차이가 나타나지 않은 반면 Beclin-1이 노화 쥐의 plantaris 근육에서 유의하게 증가[20] 되기도 또는 EDL과 gastrocnemius 근육에서 감소[27]된 것으로 보고되었다. 현재 이러한 불일치한 결과에 대한 원인은 불분명하다. Beclin-1과 달리 Bnip3, Lamp1, HDAC6와 같은 초기 자가포식체 형성 이후의 과정들에 관련된 자가포식 인자들은 젊은 쥐의 골격근에 비해 노화된 골격근에서 유의하게 감소되어 있었다(Fig. 4A, C-E). 이 결과는 앞에서 이미 제시했던 즉, 노화된 골격근에서 자가포식은 초기과정보다는 그 이후의 해결 과정의 손상에 의해 자가포식의 유동이 감소했을 것이라는 제안을 다시 보여준다.
본 연구에서 p62는 colchicine 처치에 의해 증가되지 않았고 통계적으로 유의하지 않았지만 젊은 쥐에 비해 노화 쥐의 골격근에서 증가된 양상만을 보여주었다(Fig. 3A, G). 또한 8주 수영훈련에 의해서도 뚜렷한 변화가 생기지 않았다. p62는 LC3 II에 비해 그렇게 좋은 자가포식 지표는 아닌 듯하다. p62는 자가포식 외에 다른 세포내 작용에 관여하는 듯하다. 예를 들어, p62는 Ras/Raf/MAPK, NFκB, 그리고 항산화와 종양형성에 관여한다[28].
현재 지구성 운동 훈련이 노화에 의해 변화된 골격근의 자가포식 유동에 간섭(intervention)을 일으키는 가에 대한 연구는 그리 많지 않으며 그 간섭효과 또한 명확하지 않다. 최근에 보고된 연구[29,30]와 비슷하게, 본 연구에서도 8주 수영훈련이 젊은 쥐의 골격근의 자가포식을 유의하게 증가시켰다. 젊은 쥐의 sed+col 그룹에 비해 exe+col 그룹의 LC3 II 단백질 수준이 ~100% 증가되었다(Fig. 3A,F). LC3 II/LC3 I 전환율 또한 8주 수영 훈련에 의해 젊은 쥐의 골격근에서 유의하게 증가하였다(Fig. 3A,E). 8주 수영 훈련이 젊은 쥐의 골격근의 자가포식을 증가시킨 반면 노화 쥐의 골격근에서 LC3 II 단백질 수준을 증가시키지 못하였다(Fig. 3A, F). 그리고 LC3 II/LC3 I 전환율 또한 노화 쥐의 골격근에서 변화가 나타나지 않았다(Fig. 3A, E). 젊은 쥐에 비해 노화 쥐의 골격근에서 지구성 운동 훈련에 반응하여 자가포식 유동이 증가하지 않았다. 노화 쥐의 골격근에서 자가포식 유동이 손상되어 있음을 짐작해 볼 수 있다. 이러한 이유 때문에 노화된 골격근에서 지구성 운동 훈련에 반응하여 골격근의 적응 능력이 감소된 것은 자가포식 유동의 손상에 의한 것으로 볼 수 있다. 자가포식은 골격근의 항상성을 유지시키며 근섬유를 정상적인 상태로 유지시켜 근육의 질량을 유지하는 데 매우 중요한 역할을 한다[30,31]. 따라서 자가포식의 손상은 근섬유를 약화시키고 근질량의 감소와 비정상적인 미토콘드리아에 의해 근육의 장애를 일으킬 수 있다[32]고 하였다. 노화 쥐는 아니지만 선행연구에서 자가포식이 근육 기능에 중요한 역할을 한다는 것을 유전자 변형 마우스 모델을 통해 보여주고 있다. Atg7 knockout (Atg7-/-) 마우스와 Atg5가 결핍된(Atg5+/-) 마우스에서 근질량, 힘 생성 그리고 미토콘드리아 기능의 감소가 나타났다[32,33]. 또 다른 유전자 변형 마우스 모델인 Atg6가 결핍된(Atg6-/+) 마우스는 골격근의 미토콘드리아의 생성(mitochondrial biogenesis)과 모세혈관 생성(angiogenesis)의 손상에 의해 지구성 운동 훈련에 반응하여 지구력을 향상시키지 못하였다[30]. 따라서 골격근의 자가포식은 지속적인 운동 훈련에 반응하여 근육의 적응기전에 필수적인 요인임이 틀림없는 것 같다. 8주 수영훈련은 젊은 쥐의 골격근에서 자가포식과 관련된 단백질, Beclin-1, Bnip3, Lamp1, HDAC6을 유의하게 증가시켰지만 노화 쥐의 골격근에서는 이들 단백질들의 미미한 변화만이 발견되었다. 젊은 쥐의 골격근에서 발견된 지구성 운동 훈련에 의한 자가포식의 향상 효과가 노화된 골격근에서는 나타나지 않았다. 지구성 운동 훈련에 반응하여 노화 쥐의 골격근에서 자가포식의 유동이 감소된 이유는 미토콘드리아 분해, mitophagy (Bnip3), 리소솜 생성(Lamp1), 미세소관(microtubule) 합성(HDAC6)과 같은 자가포식 활성을 촉진시키는 단백질 수준의 감소에 의한 것일 수 있다.
신체적인 운동은 건강에 많은 이점을 준다. 예를 들어, 수명 연장, 심혈관계 질환, 당뇨병, 암과 퇴행성 신경질환 등의 예방과 완화 등이 포함된다. 이러한 질병은 세포 내의 단백질과 세포소기관, 세포내 독성물질들을 제거시켜주는 능력이나 기능의 상실에 의해 생길 수 있다는 많은 증거들이 있다. 자가포식 활성화의 감소는 이러한 질병의 원인과 직접적으로 관련이 있다[26]. 최근의 연구에 의하면 운동은 자가포식을 활성화시키고 당뇨 질환이나 치매와 같은 퇴행성 신경질환을 완화시키기도 한다고 하였다[34]. 본 연구에서 지속적인 운동이 노화에 의해 감소된 골격근의 자가포식 유동을 증가시킬 수 있을 것으로 기대하였지만 노화된 골격근에서 자가포식을 향상시키지 못하였다. 현재 이러한 차이는 정확히 알 수 없지만, 노화된 골격근에서 운동에 의한 자가포식의 미미한 변화는 이 실험에서 사용한 운동 프로그램이 하나의 원인일 수 있다. 본 연구에서 사용한 운동 형태(수영 훈련) 또는 수영 훈련에 의한 낮은 강도의 신체활동이 노화된 근육에서 자가포식의 향상을 제한시켰을 가능성이 있다. 또는 훈련 기간이 원인이 될 수 있다. 8주 운동 기간은 젊은 쥐에게는 충분하지만 노화 쥐의 골격근에서 자가포식을 향상시키는 데 충분하지 않을 수 있다. 근섬유 유형에 의한 결과일 수 있다. 본 연구에서 사용한 상완삼두근(triceps muscle)은 대부분이 해당과정적(glycolytic) Type I 근섬유로 노화에 의해 Type I 섬유의 손상이 더욱 심하였기 때문에 노화된 골격근에서 자가포식의 유동의 변화를 제한시켰을지 모르겠다. 자가포식의 작은 변화에도 위에서 언급한 질환들을 예방하고 완화시킬 수 있는 능력은 향상시킬 수 있는 가능성은 있을 것 같다. 본 연구의 저자들은 차 후 연구에서 다른 운동 유형, 예를 들어, 트레드밀 달리기 운동, 또는 운동 강도와 운동 기간의 증가, Type I 섬유와 Type II 섬유를 동시에 사용하는 실험 등을 통해 노화된 골격근에서 손상된 자가포식 유동을 변화를 확인시켜줄 수 있는 더욱 자세한 연구가 필요하다고 생각한다. 또한 노화된 골격근에서 생체 자가포식 유동 분석법을 사용하여 운동이 노화와 관련된 질병이나 현상(예, 노인성 근감소증)을 개선시키는가를 확인해주는 연구 또한 중요한 가치가 있을 것 같다.
결론
본 연구는 노화된 골격근에서 자가포식 유동(autophagic flux)을 조사하였고 8주 수영훈련이 노화에 의해 변화된 골격근의 자가포식의 유동에 미치는 영향을 조사하였다.
젊은 쥐에 비해 노화 쥐의 골격근의 자가포식은 유의하게 감소되었다. 8주 수영훈련이 젊은 쥐의 골격근에서 자가포식의 유동을 증가시켰지만 노화 쥐의 골격근에서 자가포식 유동을 변화시키지 못하였다. 젊은 쥐에 비해 노화 쥐의 골격근에서 자가포식의 유동의 감소와 지구성 운동 훈련에 반응하여 손상된 자가포식의 유동의 증가는 자가포식의 개시(initiation)라기보다는 자가포식의 해결(resolution) 과정들의 원인에 의한 것으로 보여진다.