


INTRODUCTION
Vascular aging is an independent risk factor for the onset and progression of vascular diseases, such as atherosclerosis and hypertension [1–3]. A major mechanism of vascular aging and its pathological contribution to the functional deterioration is the accumulation of EC senescence, which is defined as an irreversible loss of cell proliferation capacity [3–5]. EC senescence can be induced by a number of factors implicated in the vascular pathologies such as oxidative stress [1,6]. Excessive production of reactive oxygen species generated from intracellular or extracellular sources accelerates the development of premature endothelial senescence [1,3] and is implicated in the pathogenesis of atherosclerosis [7]. Therefore, finding strategies for the protection of vasculatures against oxidative stress-induced premature senescence and functional impairment is believed to be a crucial step in the prevention of aging-related vascular diseases.
Aerobic exercise is known to alleviate the severity of atherosclerosis and improve vascular health [8]. The mechanism underlying these adaptive changes is believed to be governed by increased hemodynamic shear stress [9]. Aerobic exercise enhances the level of unidirectional laminar flow and diminishes disturbed flow occurring in certain arterial geometries, such as bifurcations and curvatures [10,11]. In cultured EC, LSS is known to modulate the endothelial activation [12] by suppressing the expression of inflammatory adhesion molecules [13] and to prevent EC apoptosis, which is a known feature of atherosclerosis [14]. In spite of the accumulating evidence for the beneficial effects of LSS on the maintenance of vascular homeostasis, our understanding of the protective role of LSS against vascular senescence is largely limited.
Sirtuins, the NAD+-dependent histone deacetylases, are known to affect multiple pathways to modulate metabolic controls and to extend the life span of a wide variety of organisms [15,16]. Mammalian sirtuin 1 (SIRT1) regulates cell metabolism, the cell cycle, and apoptosis in ECs [17]. More importantly, SIRT1 has been recognized as a key regulator for the maintenance of vascular function and the prevention of endothelial senescence [18,19]. In a previous study, the direct contribution of enhanced LSS has been shown to cause upregulation of SIRT1 expression [20].
While multiple factors play a role in bringing about the salutary effect of aerobic exercise on the maintenance of vascular homeostasis, it was hypothesized here that the unidirectional LSS is a prominent cellular mechanism responsible for prevention against vascular senescence. Thus, the aim of this study was to determine the protective effects of LSS against the oxidative stress-induced premature endothelial senescence and to elucidate its underlying mechanism.
METHODS
1. Animals
Twenty C57BL/6J mice were randomly assigned to either sedentary (SED) (n=10) or voluntary wheel running exercise (VW) (n=10) groups. Mice in the VW group were individually housed in a ratsized cage with a metal wheel with a diameter of 11.5 cm (Prevue) fitted with digital magnetic counter. Mice in the SED group were singly housed in the same ratsized cage without the running wheel. All mice were given water and food (Purina chow) ad libitum, as previously described [21]. The animal study was carried out by procedures approved by the Temple University Institutional Animal Care and Use Committee (Permit No. 4503). All sacrifices were performed under isoflurane anesthesia.
2. Carotid artery partial ligation
Carotid artery partial ligation surgery was performed to determine the importance of laminar flow on the maintenance of vascular homeostasis compared to disturbed flow. The partial ligation surgergy on the left carotid arteries (LCA) of all twenty C57BL/6J male mice at 10 weeks of age. Partial ligation of LCA was carried out as previously described [22,23]. Briefly, anesthesia was induced by an intraperitoneal injection of a xylazine (10 mg/kg) and ketamine (80 mg/kg) mixture. the epilated area was disinfected with betadine, and a ventral midline incision (4-5 mm) was made in the neck. the LCA was exposed by a blunt dissection. Three of four caudal branches of the LCA, such as the left external carotid (ECA), internal carotid (ICA), and occipital artery (OA), were surgically ligated with 6-0 silk suture leaving the superior thyroid artery (STA) as the only outflow. On the other hand, the right carotid artery (RCA) of the same mouse was left intact as the control. Then, the incision was closed with Tissue-Mend (Veterinary Product Laboratories). Mice were monitored until recovery in a chamber on a heating pad following surgery. After 1 week of recovery, ten randomly selected mice out of twenty in total were applied to VW running for a further 3 weeks.
3. High-resolution ultrasound measurements
All ultrasound measurements were taken using a VEVO 770 high-resolution in vivo micro imaging ultrasound system with a 30-MHz mouse probe (Visualsonics). A week after partial ligation surgeries, a high-resolution ultrasound measurement was performed. Mice were anesthetized with inhaled isoflurane, and body temperature was maintained on a heated stage for the duration of observation. Levels of anesthesia, heart rate, temperature, and respirations were continuously monitored. Pulse Wave Doppler mode was used at the midpoint of the common carotid arteries for measuring flow direction and velocity.
4. Cell culture
Human umbilical vein endothelial cell (HUVEC) lines from Lonza were grown in M199 medium supplemented with 20% fetal bovine serum and endothelial cell growth supplement (Sigma-Aldrich no. E2759), and maintained at 37°C in a 5% CO2 atmosphere. All experiments with HUVEC were conducted between passages 3 and 5. For shear experiments, HUVECs, grown to 90-100% confluence, were replaced with shear media and exposed to the physiological levels of shear stress for various times using a cone and plate viscometer (0.5° cone angle), which was placed inside of a CO2 cell incubator as previously described [12,21,24]. The shear media was M199 supplemented with 2% fetal bovine serum and endothelial cell growth supplement. Immediately after shear application was over, HUVECs were treated with 100 µM H2 O2 diluted in M199 complete culture medium for 1 hour. Then, HUVECs were trypsinized, reseeded onto gelatin-coated 100mm dish, 6-well plates, 12-well plates, or matrigel-coated 96-well plates according to the experimental purposes and cultured in a normal complete growth medium, containing above compounds, for a further 3 consecutive days. For the inhibition of SIRT1, HUVECs were exposed to 30 to 100 µM concentrations of sirtinol (Calbiochem no. 566320), which is a cell permeable 2-hydroxy-1-napthaldehyde derivative and a chemical inhibitor of NAD+-dependent protein deacetylases of sirtuins.
5. Senescence-associated β-galactosidase staining
The proportion of Senescence-Associated β-Galactosidase (SA-β-gal) positive cells in cultured HUVECs or carotid arteries isolated from C57BL/6J mice were determined as described previously [25]. Briefly, cultured cells or vessel tissues were fixed in a fixative solution containing 2% formaldehyde and 0.2% glutaraldehyde for 15 minutes after washing with PBS. Cells or vessel tissues were incubated overnight at 37°C in a dry incubator in a β-Galactosidase staining solution, which contains 1 mg/mL X-gal in N-N-dimethylformamide (DMF), 40 mM citric acid/ sodium phosphate (pH 6.0), 150 mM NaCl, 2 mM MgCl2, 5 mM potassium ferrocyanide, and 5 mM potassium ferricyanide. The proportion of SA-β-gal positive cells was determined by counting the number of blue-stained cells out of the total number of cells in the same field of 200× total magnification (Axiovert 40 CFL, Zeiss).
6. Western blotting and antibodies
Antibodies were from the following sources: rabbit monoclonal anti-SIRT1 (Cell signaling no. 2496), rabbit monoclonal anti-p21 (Cell signaling no. 2947), mouse monoclonal anti-p53 (Santa Cruz no. sc-126), and rabbit monoclonal anti-p16 (Abcam no. ab51243) antibodies. Mouse monoclonal anti-α-Tubulin (Sigma no. T9026) and mouse monoclonal anti-β-Actin (Sigma no. A1978) were used as the internal controls.
ECs were homogenized in ice-cold RIPA lysis buffer with protease inhibitor (Roche no. 11836153001). Proteins from cell lysates were resolved by Tris-glycine SDS PAGE and transferred to Immobilon-P PVDF membrane for standard ECL Western blotting. Total protein expression was detected by chemiluminescence. Band densitometry analyses were completed using Image J software (National Institute of Health), to direct comparisons between experimental sets. The values calculated by Image J were arbitrary units. The densities of the selected protein bands were expressed relative to the densities of the internal control genes.
RESULTS
1. Partial ligation-induced disturbed flow and vascular senescence
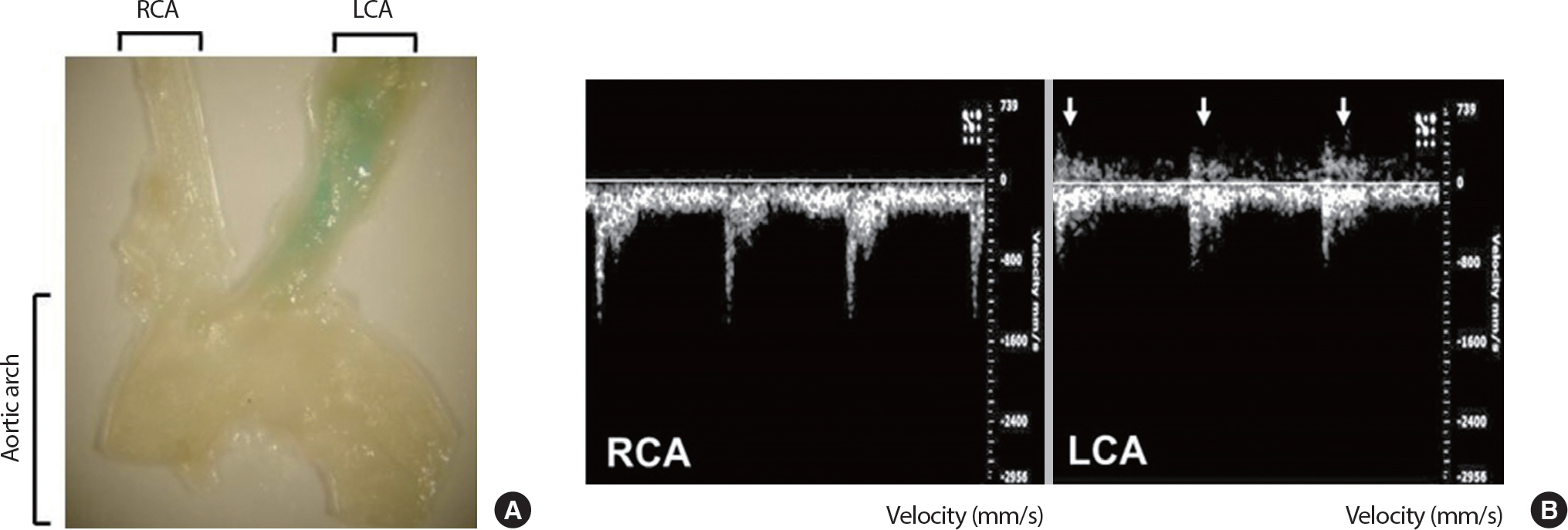
To investigate the negative effects of disturbed flow on in vivo vascular senescence compared to the positive effects of laminar flow for the maintenance of vascular homeostasis, three of four caudal branches of the LCA, such as the ECA, ICA, and OA, were surgically ligated leaving the STA as the only outflow. On the other hand, the RCA of the same mouse was left intact as the control. Flow velocity and direction of the LCA and RCA were determined by a high-resolution in vivo micro imaging ultrasound system. Results of the ultrasound showed the partial ligation-induced flow reversal in the ranges of 350 to 500 mm/s with reduced velocity to the unidirectional flow in the LCA (Fig. 1B). After 3 weeks of VW or SED after partial ligation surgery and a week recovery, mice were sacrificed and their carotid arteries were isolated for SA-β-gal enface staining analysis. SA-β-gal staining was identified only at the partially-ligated LCA of VW mice, while the LCA of SED mice and RCA from both SED and VW mice were not stained. This result represents the partial ligation and VW running-induced promoted disturbed flow in LCA leads to the vascular senescence, while laminar flow keeps the RCA intact (Fig. 1A). The average running distance of LCA partial ligated VW mice was 5.51 km/day and the body weight was significantly different in between groups and time points (pre-SED 25.39±0.72 g, post-SED 25.83±0.61 g, pre-VW 25.54±0.40 g, post-VW 25.96±0.33 g) demonstrating their behaviors, such as activity and food intake, were not affected by the ligation surgery.
Fig. 1.
Fig. 1.Partial ligation-induced promoted disturbed flow accelerates vascular senescence. Senescence-associated β-galactosidase (SA-β-gal) enface staining analysis in mouse coronary arteries after 4 weeks of LCA partial-ligation surgery and voluntary running (A). Echocardiography analysis 1-week after LCA partial-ligation surgery (B). Arrows indicate flow reversal. LCA, left carotid artery; RCA, right carotid artery; ECA, external carotid artery; ICA, internal carotid artery; OA, occipital artery; STA, superior thyroid artery.
2. SIRT1 and oxidative stress-induced premature endothelial senescence
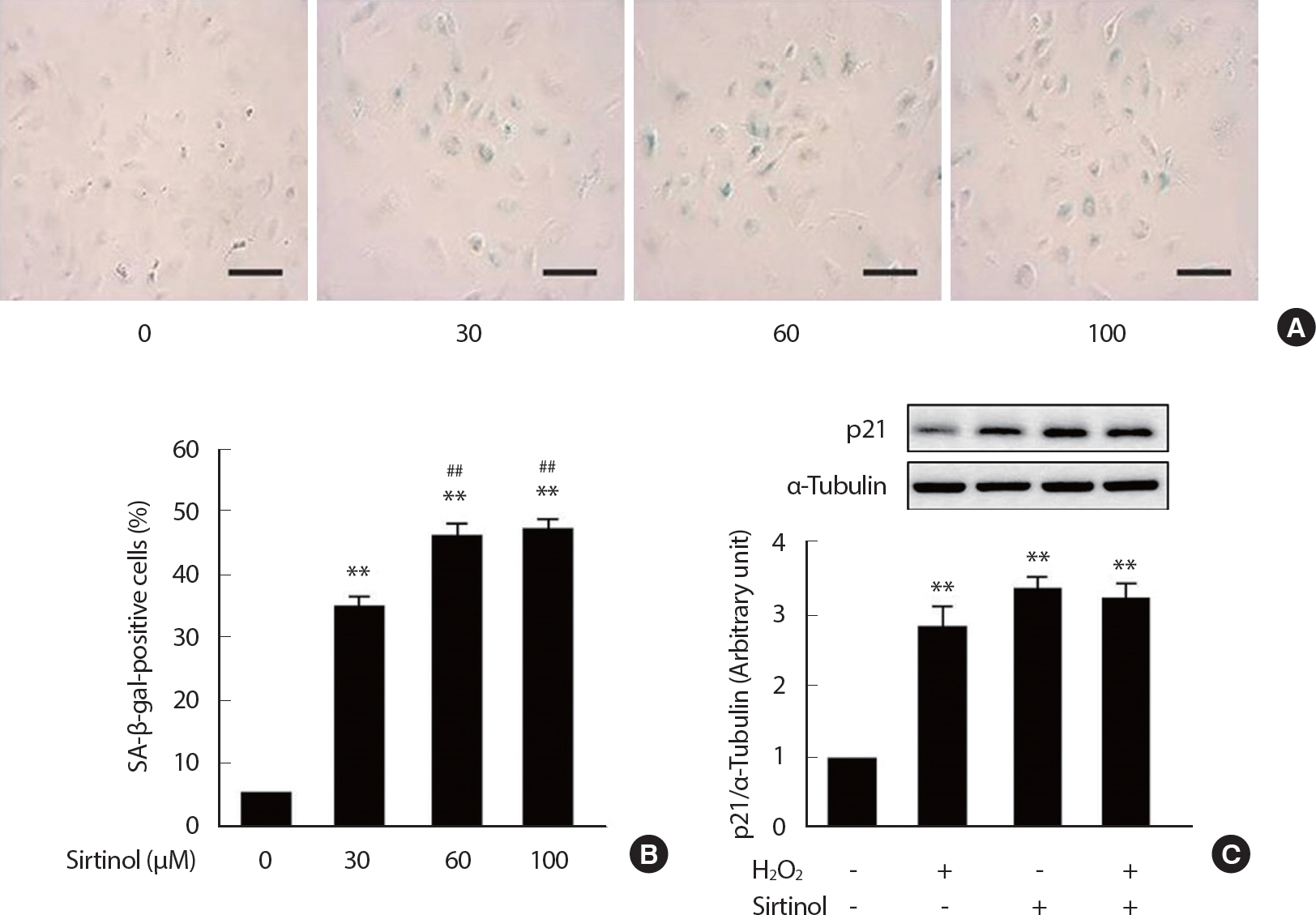
To investigate the role of SIRT1 in the regulation of EC senescence, SIRT1 was inhibited by the treatment of sirtinol, a specific chemical inhibitor of sirtuins, for 24 hours in concentrations of 30, 60, and 100 μM. Then, sirtinol-treated HUVECs were cultured in complete growth media for 3 consecutive days. The proportion of SA-β-gal-positive cells was dramatically increased in sirtinol-treated ECs in a dose-dependent manner (Fig. 2A, B). More than 45% of ECs treated with sirtinol (60 μM or 100 μM) were SA-β-gal-positive, while only about 6% of vehicle-treated ECs were SA-β-gal-positive. Senescent ECs also exhibited morphological changes of enlarged and flattened shapes. Likewise, the expressions of p21, a molecular marker of cell senescence, were significantly increased in sirtinol and/or H2 O2-treated ECs (Fig. 2C). This result confirms that the role of SIRT1 in the regulation of oxidative stress-induced premature endothelial senescence.
Fig. 2.
Fig. 2.Depletion of SIRT1 induces endothelial senescence. Representative SA-β-gal staining pictures (A). The proportion of SA-β-gal-positive cells (B). p21 expression analyzed by Western blotting (C). HUVECs were treated with sirtinol in concentrations of 30, 60, and 100 μM for 24 hours or 100 μM H2 O2 for 1 hour. Then, the cells were reseeded and cultured in normal complete medium for a further 3 consecutive days. The proportion of SA-β-gal positive cells was determined by counting the number of stained cells out of the total number of cells in the same field. Cells were counted in 60 independent fields. Scale bar=100 μm. ** p<.01 compared to non-treated control; ## p<.01 compared to 30 μM sirtinol treatment.
3. Protective effects of LSS on endothelial senescence in SIRT-dependent mechanism
To determine the protective effects of LSS on H2 O2-induced premature endothelial senescence, HUVECs were pre-treated with LSS in the intensity of 20 dyne/cm2 for 12 hours followed by 5 dyne/cm2 LSS for 24 hours before the treating H2 O2. After 3 consecutive days of aging in complete growth medium, more than 50% of H2 O2-treated ECs were SA-β-gal-positive, while only about 4% of vehicle-treated ECs were SA-β-gal-positive. This result was partly reversed in LSS pre-treated ECs. In LSS pre-conditioned HUVECs, only 18% of ECs were SA-β-gal-positive. However, the effect of LSS on the alleviation of EC senescence was completely abolished (55% SA-β-gal-positive) when SIRT1 was inhibited by sirtinol treatment (Fig. 3). The expressions of p21, p16, and p53, which are the molecular markers of cellular senescence, were in accordance with the results of SA-β-gal staining (Fig. 4). In HUVECs that under-went H2 O2-induced premature senescence, p21 expression was increased by three-fold, p16 by about four-fold, and p53 by two-fold after 3 consecutive days of aging. However, the expression levels were decreased to basal level in LSS pre-conditioned ECs, while the effect of LSS disappeared in the SIRT1-inhibited condition. These results represent that LSS can partly prevent H2 O2-induced premature endothelial senescence through SIRT1-dependent mechanism.
Fig. 3.
Fig. 3.LSS inhibits oxidative stress-induced premature endothelial senescence through the SIRT1 pathway. Representative SA-β-gal staining pictures (A). The proportion of SA-β-gal-positive staining cells (B). SIRT1 expression analyzed by Western blotting. The proportion of SA-β-gal positive cells was determined by counting the number of stained cells out of the total number of cells in the same field. Cells were counted in 90 independent fields. Scale bar=100 μm. NT=Non-treated control; * p<.05 and ** p<.01 compared to NT; # p<.05 and ## p<.01 compared to H2 O2 treatment; $ p<.05 compared to H2 O2 and LSS co-treatment.

Fig. 4.
Fig. 4.LSS decreases the expressions of molecular markers of cellular senescence. p21, p16, and p53 expressions were analyzed by Western blotting (A-D). Bar graphs are results of densitometry analyses. Each column represents mean±SEM from more than 3 independent experiments. Scale bar=100 μm. NT, non-treated control; LSS, laminar shear stress. * p<.05, ** p<.01 compared to NT; # p<.05, ## p<.01 compared to H2 O2 treatment.

DISCUSSION
Endothelial senescence, an irreversible loss of cell proliferation capacity, is directly related to the functional deterioration of ECs and age-associated diseases, including hypertension and atherosclerosis [1,5,31]. The present results demonstrated that LSS provides a prominent cellular mechanism responsible for the prevention of oxidative stress-induced premature senescence in a SIRT1-dependent mechanism in vascular ECs.
We first demonstrated in the present study that the promoted disturbed flow (or flow-reversal) induced by the carotid artery partial ligation surgery combined with VW running resulted in vascular senescence, while laminar flow keeps the coronary arteries intact in the same mice (Fig. 1). This result is attributed to the repeated exposure of endothelium to enhanced flow-reversal with attenuated laminar flow. This finding is consistent with the previous studies demonstrating that the partial ligation-induced disturbed flow leads to the endothelial dysfunction and development of atherosclerosis in the carotid arteries of high-fat diet treated ApoE−/- mice [22,23]. A recent study from another group has also shown that disturbed flow promotes endothelial senescence in the aortic arch of high-fat diet treated LDLR−/- mice [27]. Our current study shows that SA-β-gal staining was identified only at the partially-ligated VW mice's LCA, while the LCA of SED mice and RCAs from both SED and VW mice were not stained. These results represent that disturbed flow induced by the partial ligation combined with voluntary running can directly induce vascular senescence. Importantly, this is the first finding which demonstrated that vascular wall shear stress itself is a crucial determinant for the regulation of vascular senescence in wild type mice.
Diverse pharmacological treatments on vascular ECs and the trials to determine their underlying mechanisms, such as eNOS and SIRT1 activation-dependent pathways, on the prevention against EC senescence and dysfunction have been studied extensively [19,28–30]. Our data, presented here, add a new methodological insight into the prevention of EC senescence and dysfunction. This study shows that unidirectional LSS is a direct and prominent cellular mechanism responsible for the protection against oxidative stress-induced premature EC senescence. In HU-VECs that underwent oxidative stress-induced premature senescence, the rate of SA-β-gal-positive cells (Fig. 3) and the expressions of p53, p21, and p16 were significantly increased (Fig. 4). Interestingly, these results were reversed by LSS pre-treatment. However, these protective effects of LSS on endothelial senescence were completely abolished by SIRT1 inhibition. These results represent that LSS pre-conditioning has protective effects against oxidative stress-induced premature endothelial senescence through a SIRT1-dependent mechanism.
Upregulation of SIRT1 has been deeply implicated in LSS. For instance, enhanced LSS increased endothelial function via SIRT1-dependent pathway in cultured ECs [20]. Furthermore, it was shown in our previous study that the increased LSS attenuates EMP production, a determinant of endothelial activation, via SIRT1 enhancement and mitochondrial biogenesis both in in vitro and in vivo studies [12]. Also, previous studies have shown that the treatment of a known SIRT1 activator, resveratrol, in cultured human vascular ECs increases the oxidative-stress resistance by scavenging H2 O2 and preventing oxidative stress-induced cell death [33,34]. The present study has shown the possibility of protective effects of LSS on the oxidative stress-induced premature endothelial senescence.
In another previous study, it was determined that SIRT1 inhibition induces premature senescence-like phenotypes in human vascular ECs [28]. SIRT1 inhibition by the treatment of sirtinol or siRNA induced cell growth arrest and increased the proportion of SA-β-gal-positive cells. Conversely, SIRT1 overexpression prevented oxidative stress-induced SA-β-gal activity in human ECs, suggesting that SIRT1 exerts protective effects against premature endothelial senescence [28]. In this regard, we also confirmed the role of SIRT1 in premature endothelial senescence. SIRT1 inhibition by the treatment of sirtinol increased the proportion of SA-β-gal-positive cells as well as the levels of p21 expressions, representing that the depletion of SIRT1 itself can be a direct contributor for endothelial senescence (Fig. 2).
CONCLUSION
The reversal of vascular aging has been a critical issue for the prevention of cardiovascular diseases. SIRT1 has been proposed as a key contributor for the inhibition of cellular senescence. The present study high-lights the role of LSS in the protection against oxidative stress-induced premature endothelial senescence in the SIRT1-dependent mechanisms. Moreover, it was confirmed in our in vivo carotid partial ligation models that vascular wall shear stress itself is a crucial determinant for the regulation of vascular senescence. Finally, it is suggested that aerobic exercise can be a potentially effective strategy for the maintenance of vascular homeostasis against oxidative stress-induced premature vascular aging.


